Please note that ASD is now largely superseded by CASH which provides the same functionality
What is ASD ?
Designed by novelbio
ASD(Alternative splicing detector) is a user-friendly software that aims to recognize and detect differential alternative splicing(AS) exons in two samples of RNA-Seq data. Since AS events always have two obvious feature in RNA-Seq data, which are altered junction reads and altered coverage of AS-exons, the algorithm of this software considers these two factors while calculating a combine P-value to investigate whether a AS event is statistically significant or not.
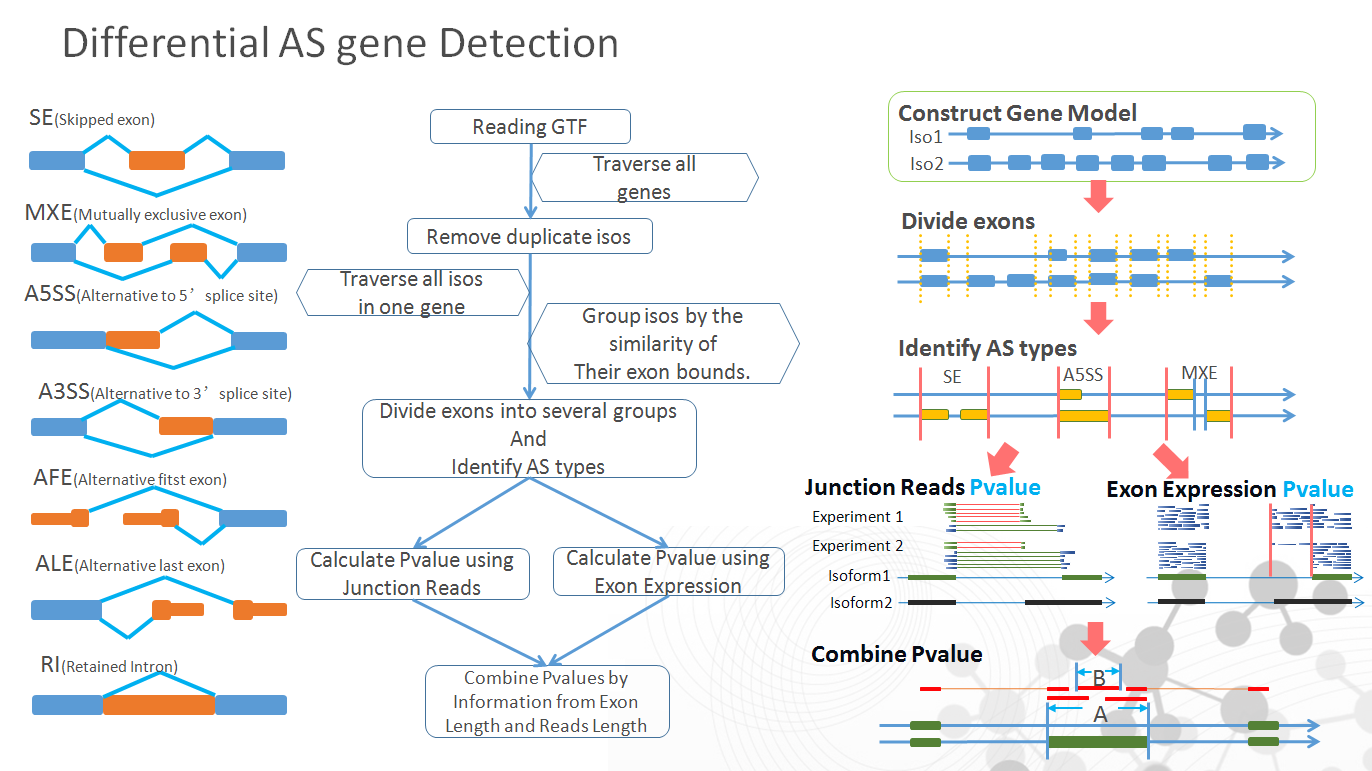
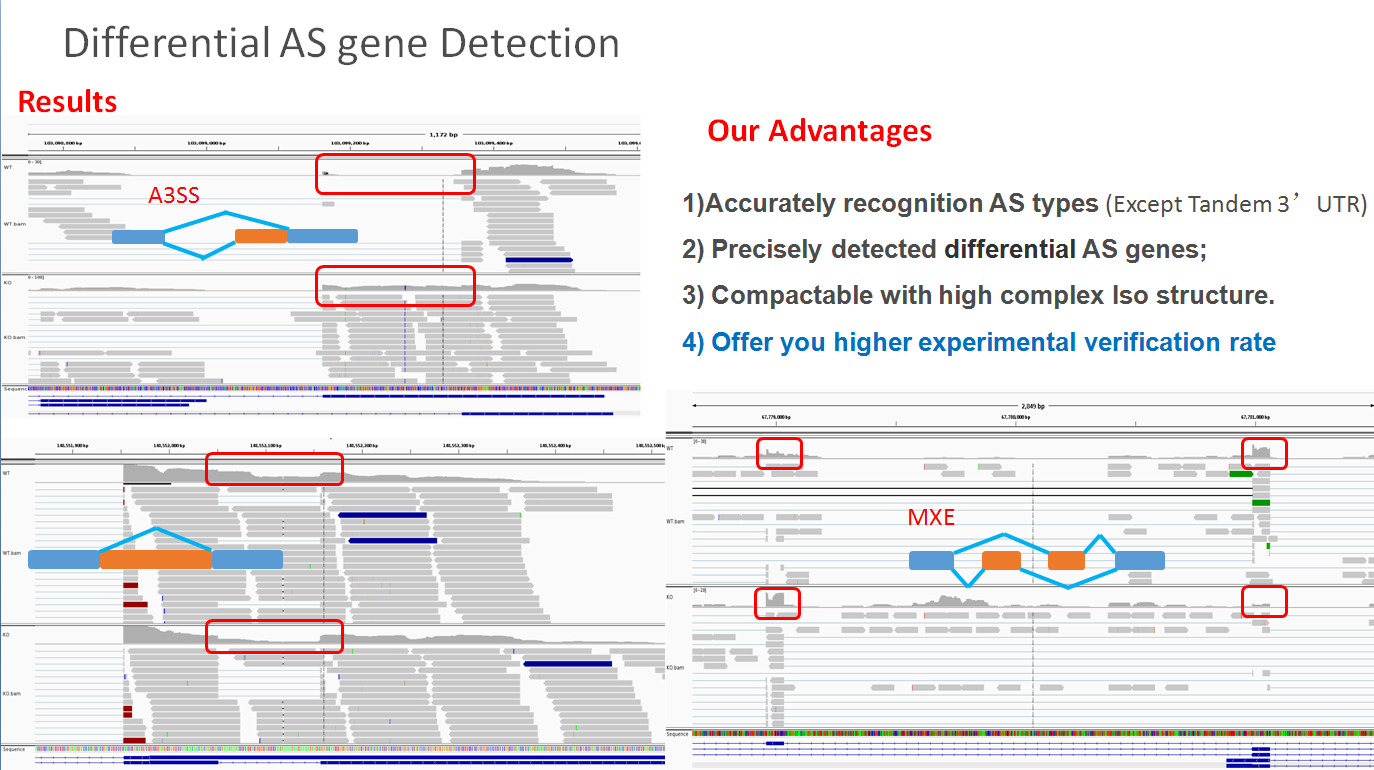
Reference:
1.
Zhou X, et al. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res. 2014 Apr;42(6):4019-30.
2.
Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors
How to use ASD ?
Installing:
ASD is a java-based software. Thus you need to install jre-1.7 first. Then you need download ASD and Assigned 755 permisssion to the ASDrun.sh if you're running ASD in linux. You would like to use the command:
sudo chmod 755 /your/path/to/ASD/ASDrun.sh
to install this
Usage: To start ASD, you just need to double click ASDrun.sh in linux or ASDrun.bat in windows. ASD need bam version file that is generated by TopHat or any other RNA-Seq maping softwares which contains the junction reads, as well as a GTF version file corresponding to the genome you use to do the mapping. Optionally, If you want to get the sequence near the splicing site, you would need to provide the genome sequence fold to ASD.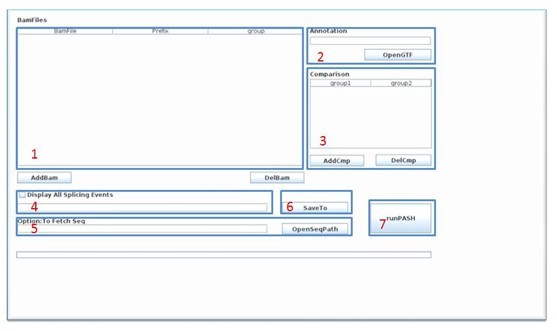
- Click button AddBam to load in the bam format files generated from the mapping software such as tophat. Fill the prefix column and group columns. Bam files come from same technical replicates should with same prefix names, and with same group names if they come form same biological replicates.
- Click OpenGTF to load in the GTF version files that records the information about reconstructed transcripts.
- Click AddCmp and then put down names of both control and treated groups, which should be consistent with those that were used at step 1.
- It is possible that there are more than one alternative splicing locations in a single gene. With "Display All Splicing Events" chosen, we could get all the alternative splicing events. With "Display All Splicing Events" not chosen,we could only get the alternative splicing events that are statistically significant.
- Click OpenSeqPath to load the folder recoding the whole genome sequences into the platform. Users need to notice that there is only one file for a single chromosome.
- Click SaveTO to set up the saving path,After the above settings, the ASD UI should like this:
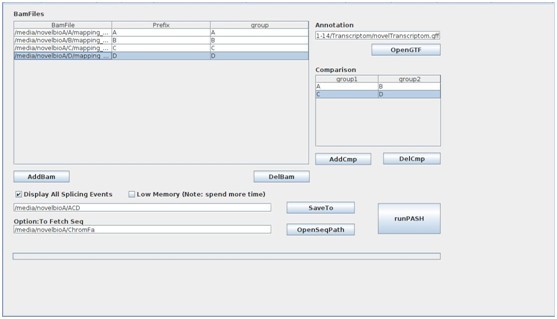
- Click runPASH to start the detection for significant alternative splicing events, and now you can have a rest and take a cup of coffee.
- Result format
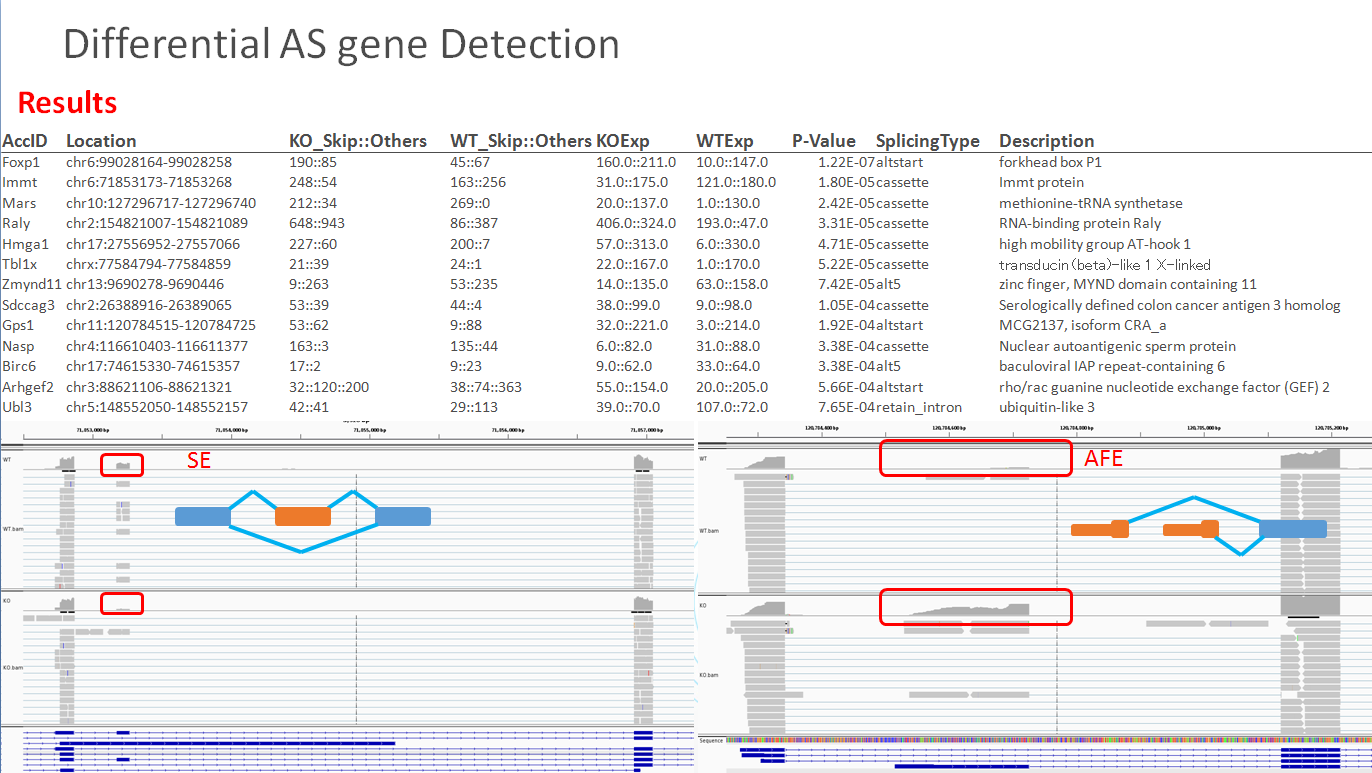
AccID Location Treat_JuncInfo Col_JuncInfo TreatCoverage ColCoverage P-Value SplicingType Description Foxp1 chr6:99028164-99028258 190::85 45::67 160.0::211.0 10.0::147.0 1.22E-07 altstart forkhead box P1 Immt chr6:71853173-71853268 248::54 163::256 31.0::175.0 121.0::180.0 1.80E-05 cassette Immt protein Mars chr10:127296717-127296740 212::34 269::0 20.0::137.0 1.0::130.0 2.42E-05 cassette methionine-tRNA synthetase Raly chr2:154821007-154821089 648::943 86::387 406.0::324.0 193.0::47.0 3.31E-05 cassette RNA-binding protein Raly Hmga1 chr17:27556952-27557066 227::60 200::7 57.0::313.0 6.0::330.0 4.71E-05 cassette high mobility group AT-hook 1 Tbl1x chrx:77584794-77584859 21::39 24::1 22.0::167.0 1.0::170.0 5.22E-05 cassette transducin (beta)-like 1 X-linked
AccID: the gene Access ID or Symbol, depends on the information provide by GTF file. Location: Splicing events happened location, canbe enter in IGV to see splicing details. Treat_JuncInfo: Junction reads number information in treat sample. Col_JuncInfo: Junction reads number information in control sample. TreatCoverage: Treatment splicing exon coverage and gene coverage ColCoverage: Control splicing exon coverage and gene coverage P-value: The significant of differential splicing event. SplicingType: Type of this alternative splicing site. Description: Desciption of this gene, depends on the information provide by GTF file. - About GTF format
the GTF file format should follow two rules:
1. No matter the strand of a gene, the location of the exon should be sorted from low to high,
2. If there is a CDS, it should be wroten after all the exons involved in.
just like the table below:Alse you can only have exon infomation without CDS information, because ASD extract the junction infomation from exon lines.chr1 novelbio exon 865535 865716 . + . gene_id "SAMD11"; transcript_id "NM_152486"; gene_name "SAMD11"; chr1 novelbio CDS 865535 865716 . + . gene_id "SAMD11"; transcript_id "NM_152486"; gene_name "SAMD11"; chr1 novelbio exon 866419 866469 . + . gene_id "SAMD11"; transcript_id "NM_152486"; gene_name "SAMD11"; chr1 novelbio CDS 866419 866469 . + . gene_id "SAMD11"; transcript_id "NM_152486"; gene_name "SAMD11"; chr1 novelbio exon 879583 880180 . - . gene_id "NOC2L"; transcript_id "NM_015658"; gene_name "NOC2L"; chr1 novelbio CDS 880077 880180 . - . gene_id "NOC2L"; transcript_id "NM_015658"; gene_name "NOC2L"; p_id "P5581"; tss_id "TSS17475"; chr1 novelbio exon 880437 880526 . - . gene_id "NOC2L"; transcript_id "NM_015658"; gene_name "NOC2L"; p_id "P5581"; tss_id "TSS17475"; chr1 novelbio CDS 880437 880526 . - . gene_id "NOC2L"; transcript_id "NM_015658"; gene_name "NOC2L"; p_id "P5581"; tss_id "TSS17475";
Which to choose?
Version 1.2
This version fix bugs and optimize the transcriptome reconstruct process. And can now support Gff3 files download from NCBI( the Human, Mouse and Rat Gff3 have already been tested).
2014-10-10
Version 1.1
This version fix bugs and add a function to reconstruct transcriptome, and can detect splicing events even not include in gff file.
2014-04-18
Version 1.0
This is the first Version!
2013-07-18