NAME
CASH - Comprehensive alternative splicing hunting
Description
CASH (Comprehensive alternative splicing hunting) is visualized and user-friendly software that aims to self-construct AS (alternative splicing) sites and detect differential AS events between samples of RNA-Seq data. CASH consists of two major stages: SpliceCons (Splice site Construction) and SpliceDiff (differential AS detection). By comprehensively reconstructing AS sites from RNA-seq data, SpliceCons increases the recognition of AS events considerably and subsequently, SpliceDiff uses two combined statistical methods to improve the detection of differential AS events. Different from our previous ASD program, we developed a new module named SpliceCons implemented into CASH and also refined the scripts to detect differential AS events. We recommend using CASH, which now replaces the original ASD version. The users can download CASH from sourceforge (https://sourceforge.net/p/cash-program).
CONTENTS
Download
Quick Start
Requirements
For CMD users
For GUI users
GTF Format
Output Format
License And Citation
Download
Download the latest CASH and example data.
Quick Start
Usage:
java -jar -Xmx10g cash.jar [options] \
--Case:prefix1 case.bam \
--Control:prefix2 Control.bam \
--GTF genes.gtf --Output outFilePrefixExample:
java -jar -Xmx10g cash.jar \
--Case:Mutation file1.bam,file2.bam \
--Control:WildType file3.bam,file4.bam \
--GTF genes.gtf --Output outFilePrefixOr GUI interface is obtained by running: java -jar -Xmx10g cash.jar --GUI
IMPORTANT: Make sure all the input bam files were sorted and indexed!Requirements
CASH requires jre1.8 or later, and at least 8GB memory for 2vs2 human samples.
For CMD users
In console, go to the directory that includes CASH, unzip CASH and cd the CASH directory as follows:
unzip CASH_v2.20.zip
cd CASH_v2.20
java -jar cash.jar --help
Required parameters
Input:Output:
--Case:prefix1 files sorted and indexed case bam files, using comma to separate replicated samples. e.g. --Case:KO /home/user/ko1.bam,/home/user/ko2.sorted.bam --Control:prefix2 files sorted and indexed control bam files, using comma to separate replicated samples.
e.g. --Control:WT /home/user/wt1.bam,/home/user/wt2.sorted.bam
Note: indexed file (bai file) can be absent if the parameter "--runSepChr" is set to be False--GTF file genes.gtf, CASH needs reference gene annotation (eg. gtf/gff file) and RNA-seq data to construct alternative splicing (AS) model within genes
--Output prefix outFilePrefixOutput directory and prefix, e.g. --Output /home/user/myresult
Options:
--MergePval A/G , Default is G. It is recommend to use the default value(G), while the results showed a poor number is more sensitive. Users can switch G to A and get more specific results
A: arithmetic weighted mean of event-centric strategy and exon-centric strategy Pvalues(more specific)
G: geometric weighted mean of event-centric strategy and exon-centric strategy Pvalues(more sensitive)--Combine True/False Default is False.
False: if here are several replications, CASH treats them as biological replicates as usual.
True: if here are several replications, CASH combines case(control) bam files to be one case(control) big bam file.--DisplayAllEvent True/False Default is True
A gene may have several AS events on different exons, CASH can display all events, or just show only one most significantly event
True: show all splicing event
False: show only one most significantly splicing event--StrandSpecific F/R/NONE Default is NONE
When the sequence library is strand specific, the parameter is used
F: first read of the pair-end reads represent the strand of the fragment, just like ion proton R: second read of the pair-end reads represent the strand of the fragment--SpliceCons True/False Default is True
SpliceCons is used to construct AS model based on RNA-seq data and reference gene annotations, leading to detection of novel AS events in the samples
True: constructing AS model based on RNA-seq data and gtf/gff files. The process needs more time
False: employing AS model inferred from gtf/gff file--JuncAllSample int Default is 25
Doesn't calculate AS event with the sum of all sample junction reads less than JuncAllSample.--JuncOneGroup int Default is 10
Doesn't calculate AS event with one group of junction reads less than JuncOneGroup.--minAnchorLen/-A int Default is 5
When counting junction reads, exon-exon junctions spanned by reads with at least this many bases on each side.--minIntronLen/-I int Default is 25
The gaps between RNA-Seq reads with length > 25bp is considered to be intron.--minJuncReadsForNewIso/-J int Default is 10
Min junction reads for reconstruct AS site.--runSepChr True/False Default is True
Due to some species (e.g. Hordeum vulgare) chromosomes with a huge length of base pairs, the java module 'htsjdk(v2.9.0)' can hardly support the index of the chromosomes and to fix the issue, we added this parameter and users can set this parameter to False, which means CASH run without index files, but it will take more memory and more computing time.--ChrRegion chrID/chrID:startPos-endPos While runSepChr is True(default), one can set this parameter and CASH will only calculate this region.You can set value as chromosome Id like "--ChrRegion chr1" or set a specific region like "--ChrRegion chr1:1-9527". --LogDebug Print debug information of CASH. For GUI users
1. Down the latest CASH and example data and unzip it into the directory that the users want.
2. Go to the directory that includes CASH, directly double click cash.bat or cd the CASH directory that you set and type 'java -jar -Xmx10000m cash.jar' --GUI in CMD command.

3. An interface exhibits as follows:
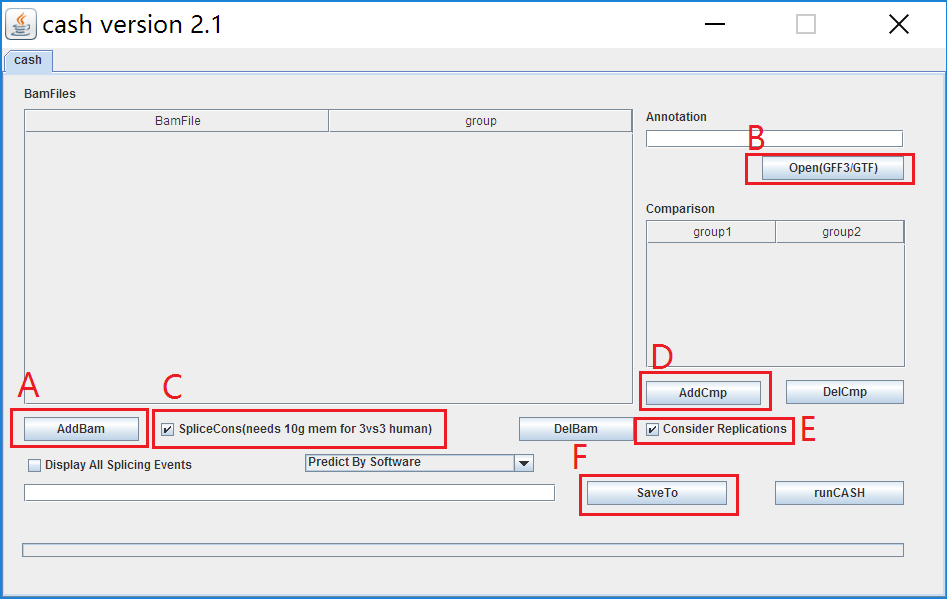
4. A) Load RNA-seq bam files: Click button AddBam to load .bam format files from your system. If the experiments have RNA-seq replications, do some edits in group column to make the control files classified into Control group (e.g. Untreated) and the test files into Test group (e.g. RNAi).
B) Load GTF or GFF3 file: Click OpenGFF3/GTF to load the file of reference gene annotations.
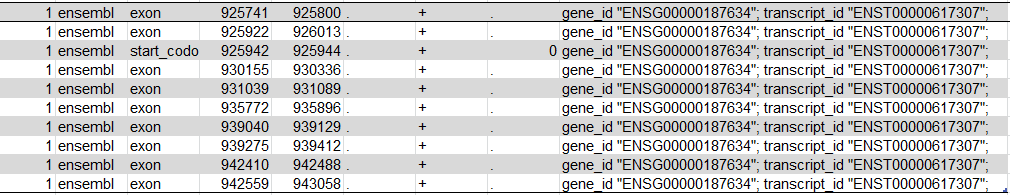
About GTF format: the GTF file format should follow two rules:
1. No matter the strand of a gene, the location of the exon should be sorted from low to high;
2. If there is a CDS, it should be wroten after all the exons involved in.
just like the table below: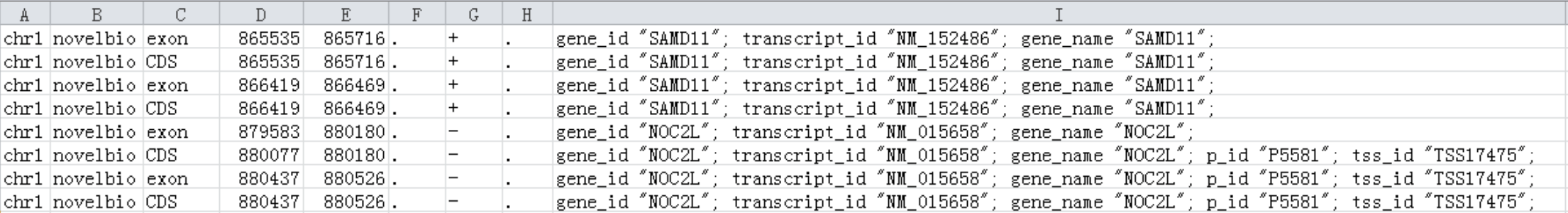
C) Construction of Alternative Splicing (AS) sites: Tick off SpliceCons to construct AS sites based on RNA-seq data and reference gene annotations, leading to detection of novel AS events in the samples. Otherwise, detection of AS events relies on reference gene annotations.
D) Groups comparison: Click AddCmp to set the two compared groups.
E) The samples with replications: Tick off Consider replications indicates CASH will treat the samples in the same group as biological replicates. However, when the sequencing depth is too low to detect AS events, we suggest the users not to tick off Consider replications, and in that way, CASH will integrate the bam files in the same group into one bam file to enhance the detection of AS events.
F) Save the results: Click SaveTo to store the analysis results.
5. An example of loading files and setting parameters.
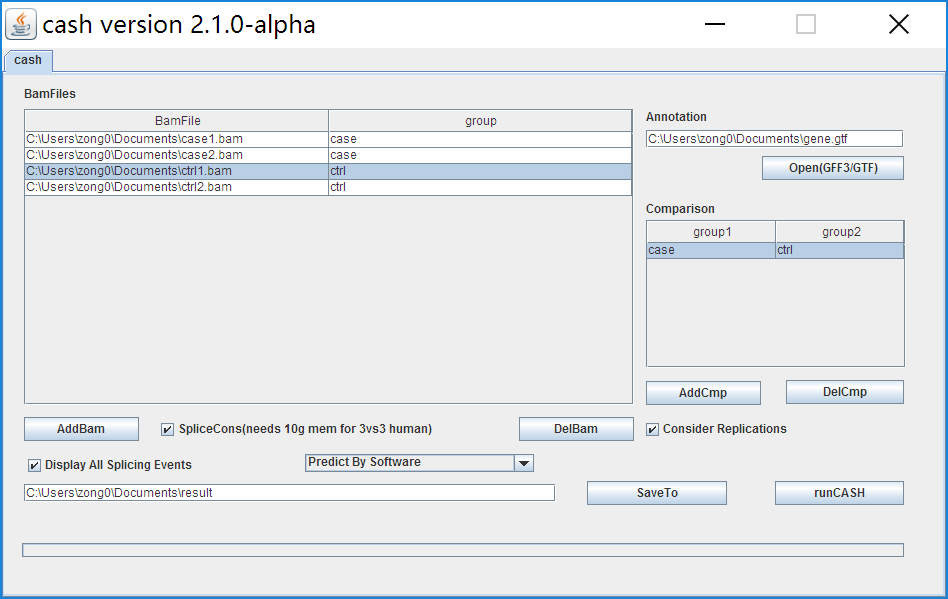
6. runCASH: When all required files are loaded, start running CASH.
7. Output files: Two output files are generated in the output directory, that is, for example, 20151220_WTvsKO_statistics.txt presents the summary of AS splicing types in the samples, and 20151220_WTvsKO.txt presents details for all of the AS events in the samples.
GTF FORMAT
About GTF format: the GTF file format should follow two rules:
1. No matter the strand of a gene, the location of the exon should be sorted from low to high,
2. If there is a CDS, it should be wroten after all the exons involved in.
just like the table below:
Output files
Two output files are generated in "examples" directory with the prefix "results", that is, results_KOvsWT_statistics.txt presents the summary of AS events between the samples, and results_KOvsWT.txt presents details for all of the AS events between the samples.
The output result as follows:
The output result detail: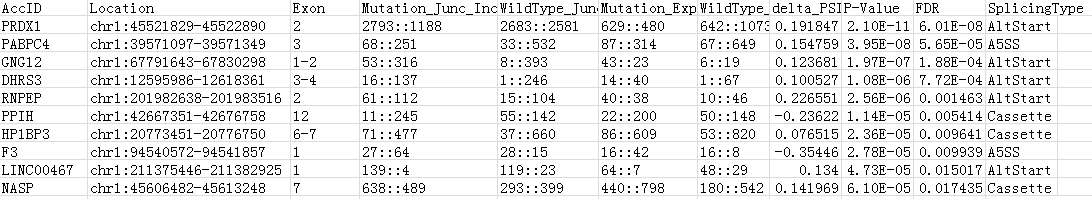
AccID Gene name Location The loci of AS events at chromosome #Exon Which exon has alternative splicing within the gene Mutation_Junc_Inclusive::Exclusive The counts indicate the expression of two types of junction reads (inclusion and exclusion junction reads) for Mutation samples WildType_Junc_Inclusive::Exclusive The counts indicate the expression of two types of junction reads (inclusion and exclusion junction reads) for WildType samples Mutation_Exp_Inclusive::Exclusive The counts indicate the sequencing depths of two type exons (inclusion and exclusion exons) for Mutation samples WildType_Exp_Inclusive::Exclusive The counts indicate the sequencing depths of two type exons (inclusion and exclusion exons) for WildType samples delta_PSI P-Value is calculated based on a weighted mean of event-centric strategy and exon-centric strategy FDR False Discovery Rate using Benjamini-Hochberg method SplicingType Types of AS event LICENSE AND CITATION
The source code of CASH is under Apache License V2.0.
CASH is free for academic usage, please cite the papers when you use it. For commercial usage, please contact Dr. Wenwu Wu (wenwu_wu@aliyun.com) or Dr. Jie Zong (zongjie@novelbio.com).1. Zhou X, Wu W, Li H, Cheng Y, Wei N, Zong J, Feng X, Xie Z, Chen D, and Manley JL et al. 2014. Transcriptome analysis of alternative splicing events regulated by SRSF10 reveals position-dependent splicing modulation. Nucleic Acids Res 42: 4019-4030.
2. Wu W, Zong J, Zhou X, Chen J, Cheng Y, Li H, Chen D, Guo Q, Zhang B, and Feng Y. 2014. Comprehensive Alternative Splicing Hunting (CASH) and its application to reveal evolution of SRSF10-regulated splicing models in vertebrates. In preparation.
cash.2.0.1 cash 29 Dec. 2015