服务热线02152235399
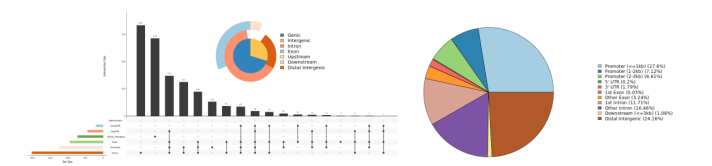
基于10X Geomics ChromiumTM动态微流控技术,利用Tn5转座酶酶切开放染色质,形成短片段,单细胞ATAC测序在表观基因组学水平上,揭示单细胞染色质的可及性问题,区分细胞异质性,获得开放染色质的位置、转录因子的结合位点、核小体的调控区域和染色质状态等信息,是单细胞表观遗传学的重要突破。

Alexandro E. Trevino et al. Cell. 2021 Sep
1.突破性改进制核手段:跳过样本消化步骤,直接冻存组织进行制核。这种方法可以大大节省时间和资源,并且提高制核效率,细胞核捕获率高达65%-80%
2.消除细胞解离偏差:消除由于消化偏好性造成的细胞解离偏差,保证制核质量远高于上机捕获要求,可以为后续的分析提供更准确的结果。
3.严格质量把控:烈冰全程进行严格的质量把控,从实验设计到分析产出提供一站式服务流程,确保客户获得高质量的数据和分析结果,并提供全面的支持和解决方案。
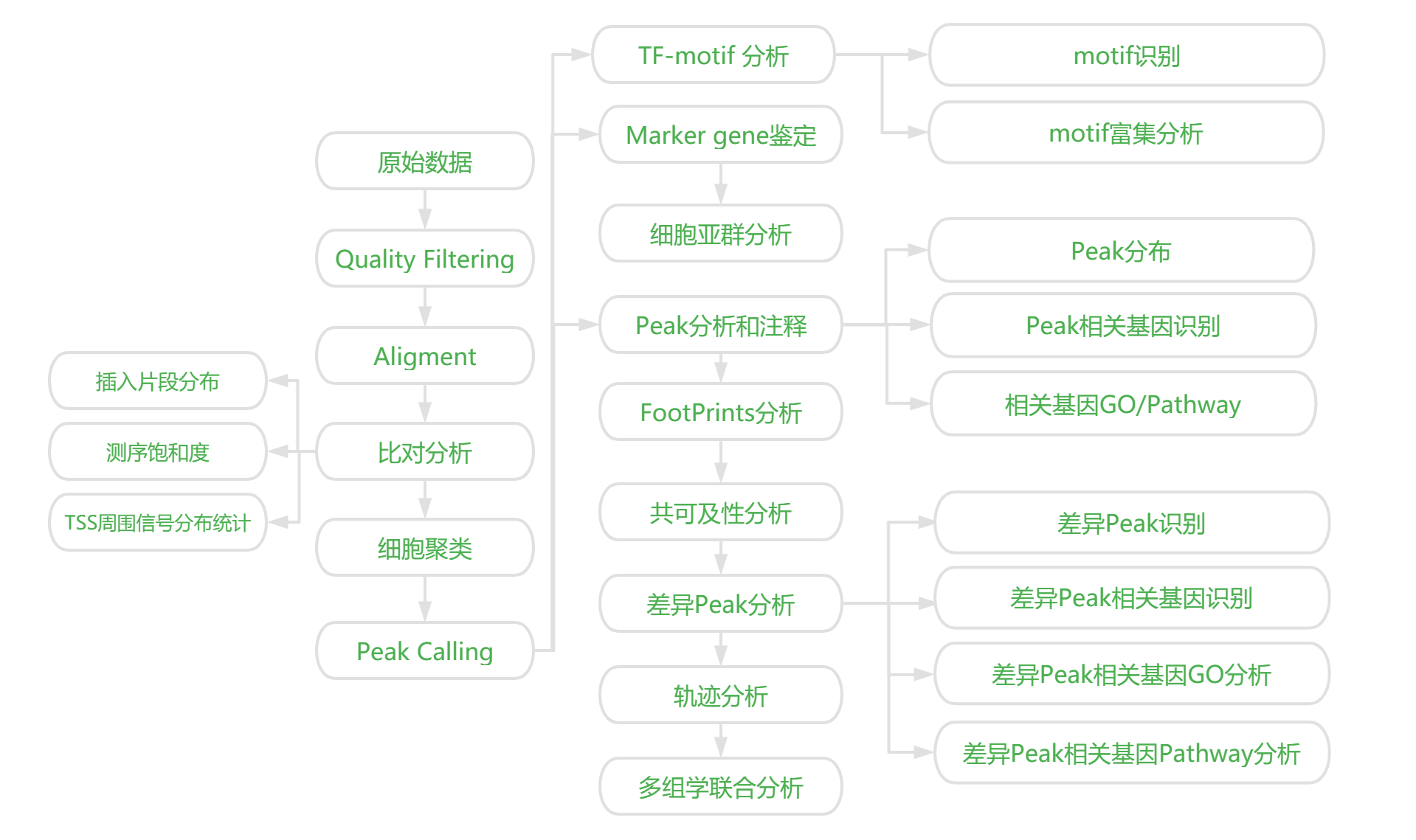
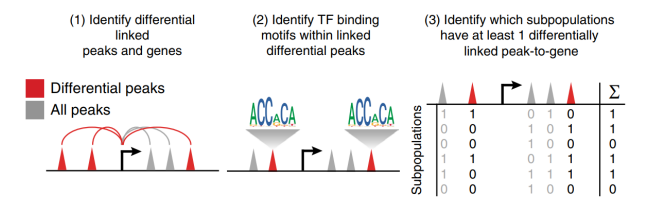
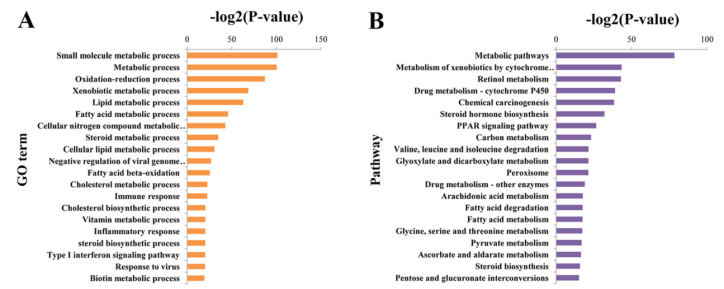
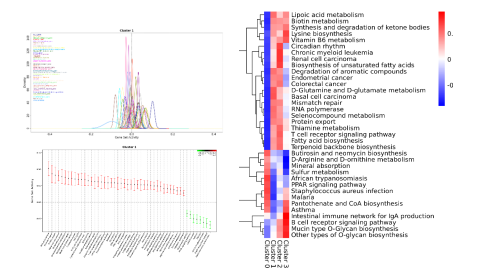
4.烈冰自主搭建分析云平台,一键实现数据分析,包括peak calling、聚类、TF-motif、细胞亚群、Marker基因和信号通路富集分析等
样本类型:
组织、血液、培养的细胞系、制备好的单细胞悬液
质量要求:
1. 细胞活性大于70%
2. 浓度为500-2000细胞/μl
3. 体积不小于200μl
4. 细胞培养基及缓冲液不能含Ca2+和Mg2+
5. 细胞体积小于40μm






客户样本--悬液制备---活性检测--活细胞富集--细胞核制备--核质控检测--单细胞核捕获--细胞/转录本标签添加--文库构建--上机测序
Jason D, et al. Cell, 2018; Wenliang Wang et al. PNAS, 2020.

Darren A. Cusanovich, et al. Cell, 2018.
Jeffrey M., et al. Nature Biotechnology, 2019.
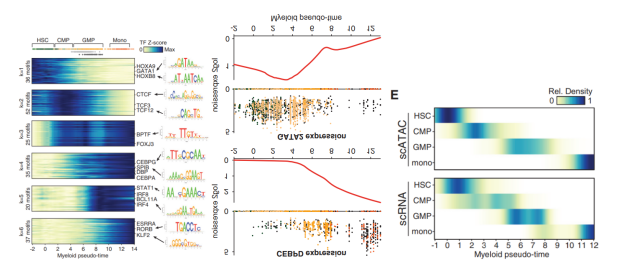
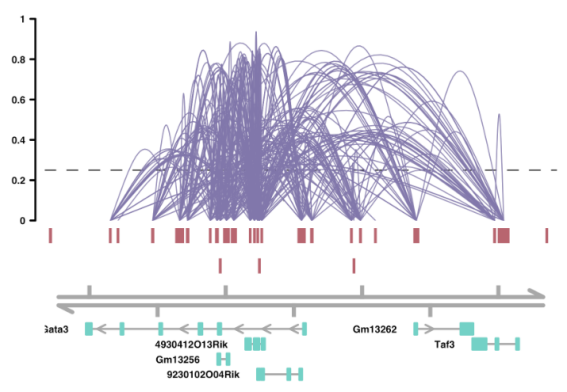
Jason D, et al. Cell, 2018.
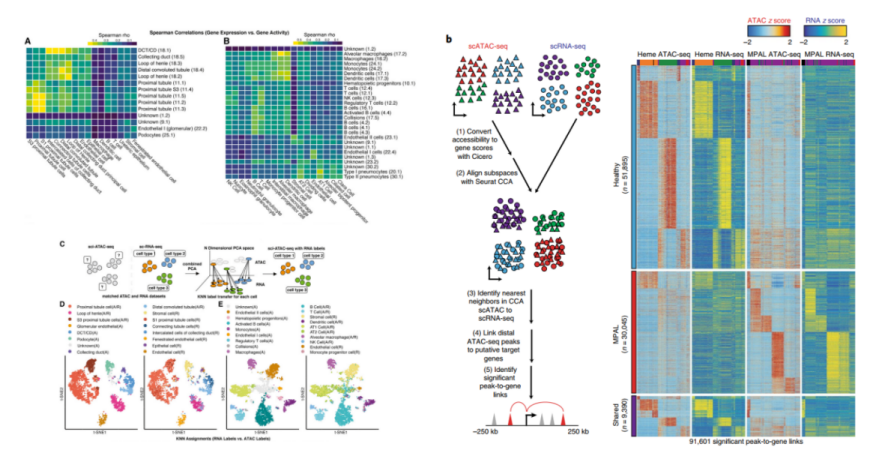
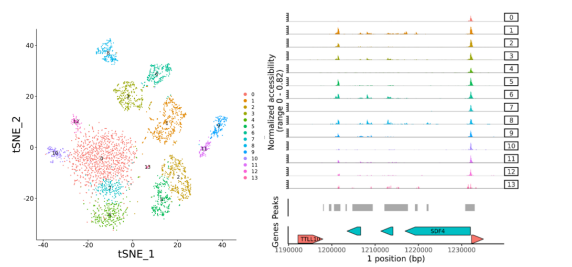
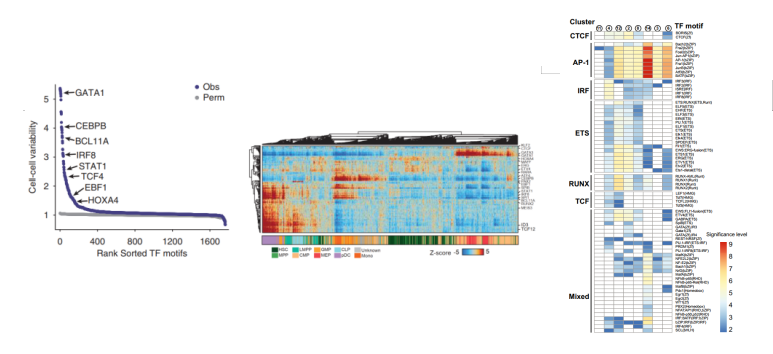
左图为单细胞RNA-seq和ATAC-seq数据整合:通过整合单细胞转录组和ATAC数据,确定ATAC细胞类型注释的可靠性与一致性。
[1] A longitudinal single-cell atlas of treatment response in pediatric AML. Cancer Cell. 2023 Dec; IF=52.9
[2] Transcription factor NFYa controls cardiomyocyte metabolism and proliferation during mouse fetal heart development. Dev Cell. 2023 Dec;IF=20.3
[3] A discrete 'early-responder' stromal-cell subtype orchestrates immunocyte recruitment to injured tissue. Nat Immunol. 2023 Dec; IF=41.0
[4] Single-cell multiomics of the human retina reveals hierarchical transcription factor collaboration in mediating cell type-specific effects of genetic variants on gene regulation. Genome Biol. 2023 Nov ; IF=25.5
[5] Single-Cell Transcriptome Atlas and Regulatory Dynamics in Developing Cotton Anthers. Adv Sci (Weinh). 2023 Jan; IF=19.8
[6] Single-cell epigenetic, transcriptional, and protein profiling of latent and active HIV-1 reservoir revealed that IKZF3 promotes HIV-1 persistence. Immunity. 2023 Nov; IF=57.7
