服务热线02152235399
转录组测序(RNA-Seq)的研究对象是特定细胞在某一功能状态下所能转录出来的所有mRNA的总和。新一代高通量测序技术能够全面快速的获得某一物种特定组织或器官在某一状态下的几乎所有转录本序列信息,从而准确地分析基因表达差异、基因结构变异、筛选分子标记(SNPs或SSR)等生命科学重要问题。
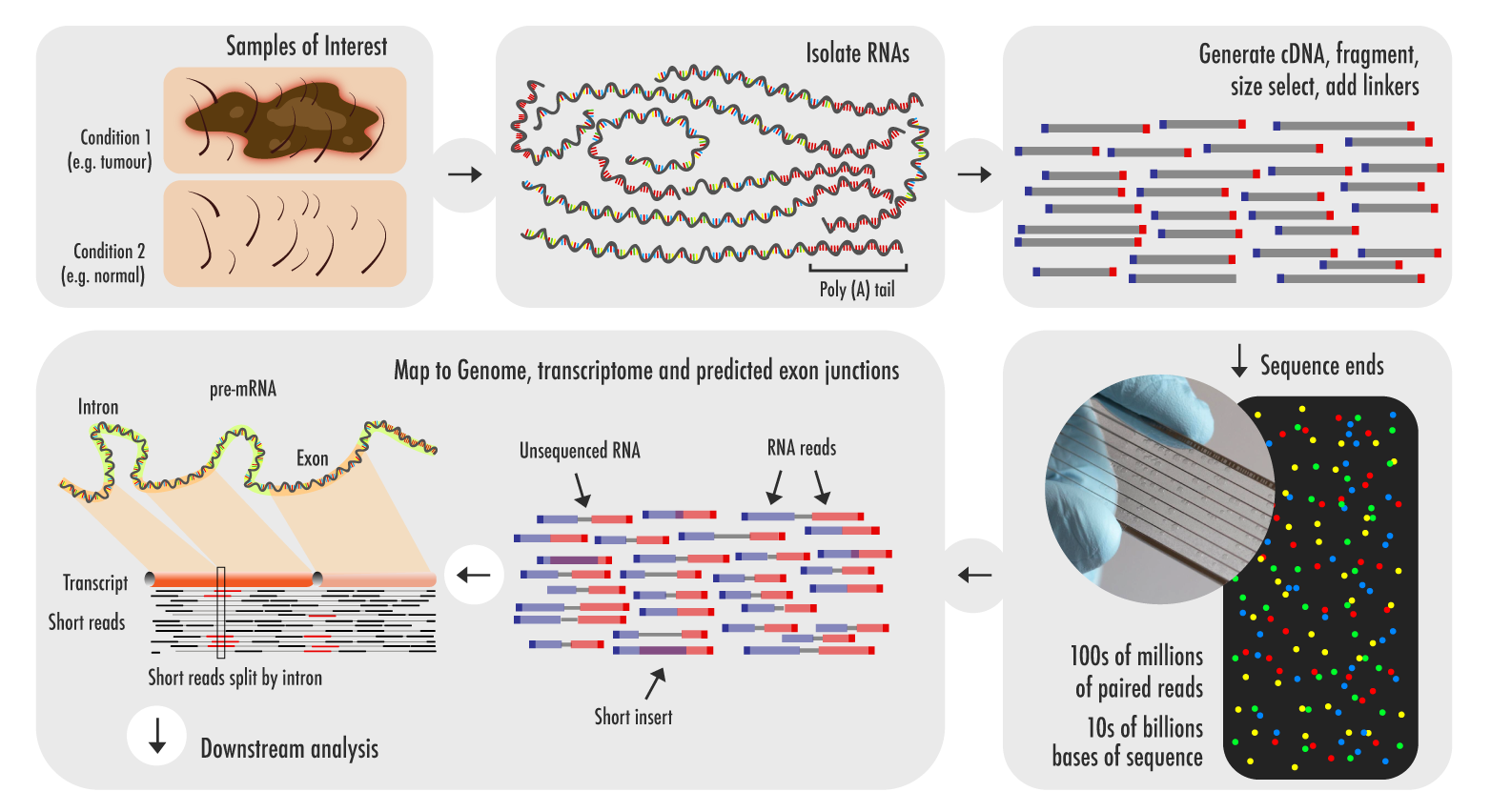
A workflow for RNA-seq
Ruairi J, Genomics Research, 2018
1. 十三年转录组测序分析经验,自主研发了多个生物学领域内认可的算法,如差异可变剪接算法ASD、CASH等,检出率和准确度超过同类软件;
2. 不依赖已有物种信息,可研究非模式物种,针对不同平台的数据,制定多套流程;
3. 烈冰自主搭建转录组分析云平台,一键实现数据分析,包括基础分析和高级数据分析
4. 整合了众多学术界公认的转录组相关数据库,从本质上提高后期分析的广度和精度,助力SCI文献300+,助力研究团队在Nature发表全球首例自闭症猴模型


(图片来源:Liu Z, et al. Nature., 2016)
组织样品:
1. 动物组织≥1g;
2. 植物组织≥2g;
3. 细胞样品≥1×106个;
4. 全血≥2mL;
5. 菌体≥106个或≥30mg。
RNA样品:
1. 样品需求量: RNA≥10 μg;
2. 样品浓度:RNA样品≥100 ng/μl;
3. 样品纯度:OD260/OD280在1.8-2.2之间,OD260/OD230≥2,28S/18S≥1,动物样品RIN≥7.0,植物样品RIN≥6.5,RNA无明显降解。





1. 客户样本:保证细胞量在106个以上,否则则需风险建库;
2. RNA提取:经典试剂盒快速提取法;
3. RNA质控:凝胶电泳质控→Nanodrop质控→Agilent2200质控;
4. 文库构建:polyA建库;
5. 上机测序:烈冰建议选择NovaSeq测序平台,双端测序,通量大,碱基精度高,且成本低,速度快。推荐数据量:6Gb。
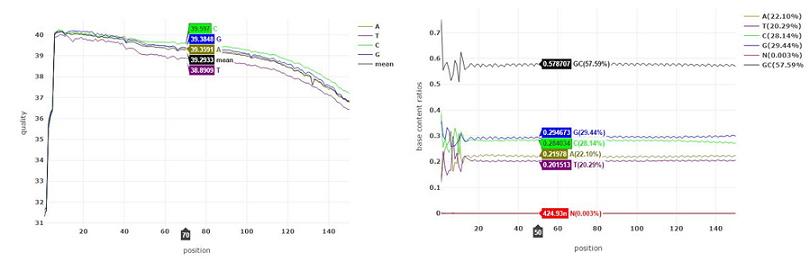
碱基质量结果图
注:左图横坐标代表碱基位点,纵坐标代表碱基质量值,不同颜色曲线代表不同碱基在每条read上的质量值;右图横坐标代表碱基位点,纵坐标代表碱基含量比值,不同颜色曲线代表不同位点各碱基含量。
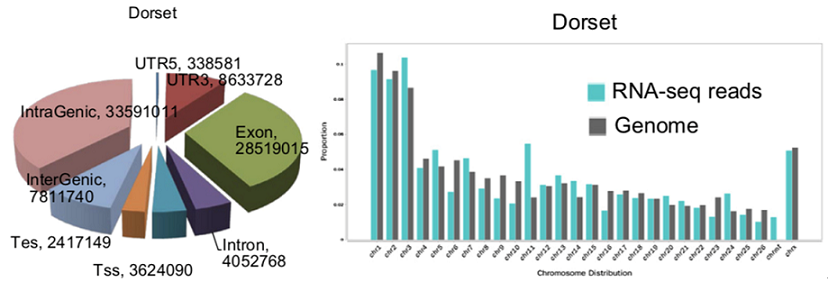
reads在基因结构和染色体上的分布情况
Miao et al., Mol Cell Endocrinol, 2015
注:左图为reads在不同基因结构(如外显子、内含子、基因间区、5’-UTR和3’-UTR)上的分布情况;右图为reads在染色体上的分布情况,横坐标表示染色体编号,纵坐标表示百分比,灰色柱子表示每条染色体上碱基数占基因组的比例,绿色柱子表示比对到染色体上reads的碱基数占基因组的比例。
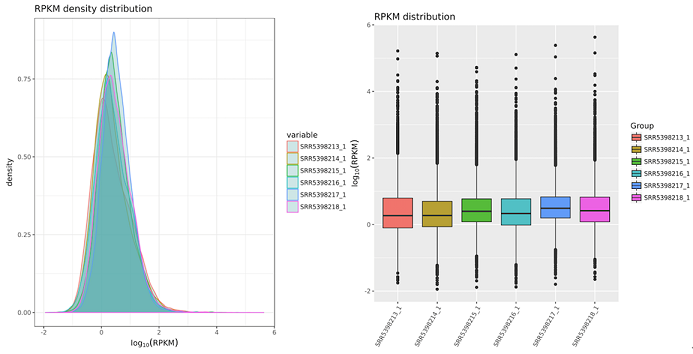
基因表达量分析
注:左图为不同样本RPKM密度图,横坐标表示log10(RPKM),纵坐标表示每个log10(RPKM)值对应的基因数占比;右图为不同样本基因表达箱线图,横坐标表示不同样本名称,纵坐标表示样本中每个基因log10(RPKM)分布情况。
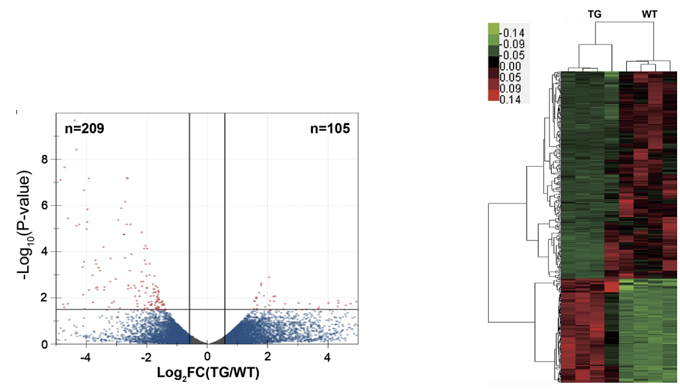
差异基因的火山图和聚类图
liu et al., Nature, 2016
注:左图为差异基因的火山图,红色表示显著差异基因,蓝色表示非显著差异基因;右图为基因表达聚类图,横坐标为样品分组,纵坐标为基因,红色表示高表达,绿色表示低表达。
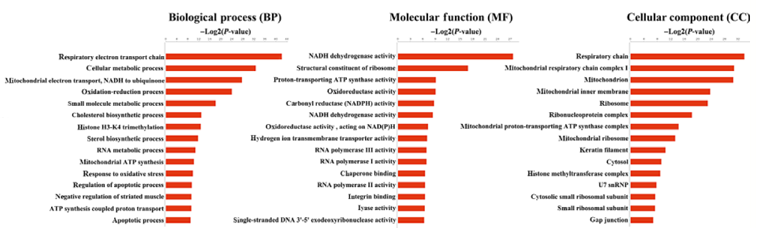
基因功能分析
He et al., Cancer Sci, 2017
注:该图从生物学进程(Biological Process, BP)、分子功能(Molecular Function, MF)和细胞组分(Cellular Component, CC)3个层面展示了差异基因显著富集的前15个功能条目。横坐标为-Log2(P-value)/-Log10(P-value),纵坐标为Go-Term条目名称。
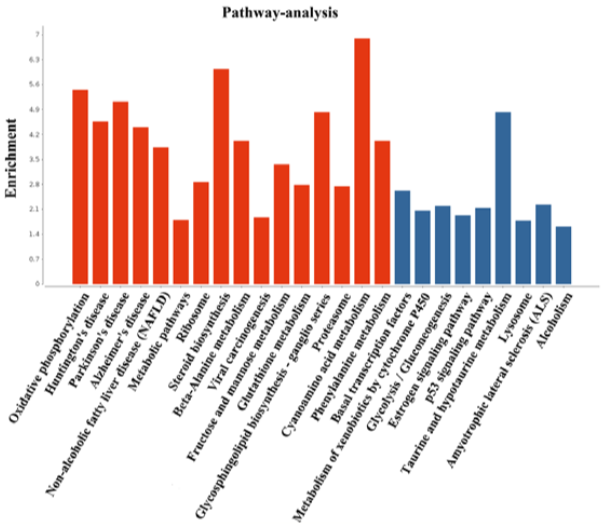
Pathway富集性分析
He et al., Cancer Sci, 2017
注:该图展示了差异基因富集的25条Pathway条目。横坐标为Pathway条目名称,纵坐标为富集度(Enrichment),红色表示显著性条目,蓝色表示非显著性条目。
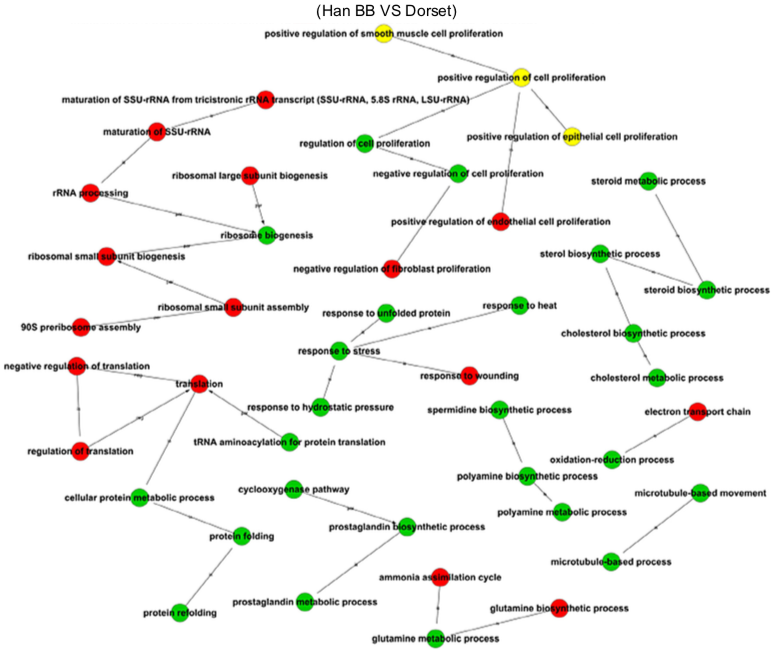
GO Tree Miao et al., Mol Cell Endocrinol, 2015
注:该图展示了差异基因显著富集的GO Terms内在从属关系。红色代表上调基因显著富集的功能条目;绿色代表下调基因显著富集的功能条目,黄色代表上调和下调基因都显著富集的功能条目。
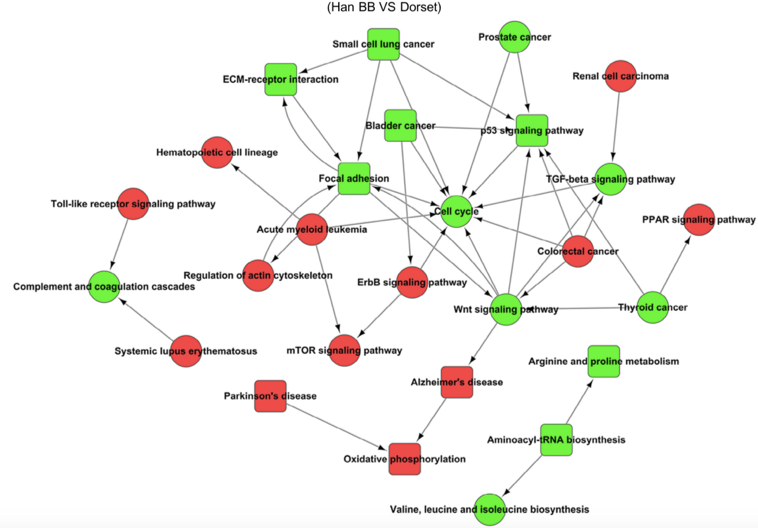
Path-Act-Network Miao et al., Mol Cell Endocrinol, 2015
注:该图展示了差异基因显著富集pathway之间的上下游调控关系。红色表示上调基因显著富集的pathway;绿色表示下调基因显著富集的pathway。
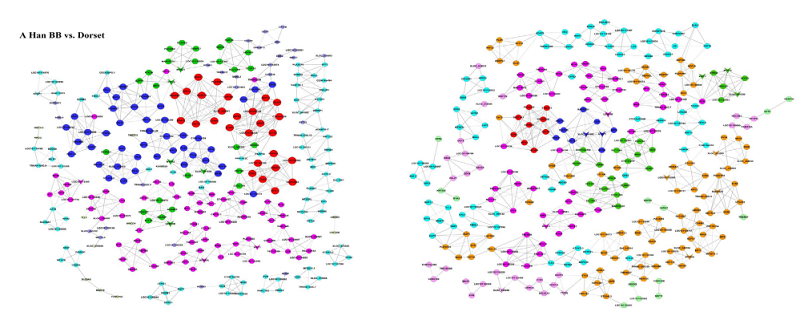
共表达网络
Miao X et al., Scientific reports, 2016
注:相同颜色的圆点表示具有相似共表达能力的基因,圆点的大小表示该基因的K-core程度。
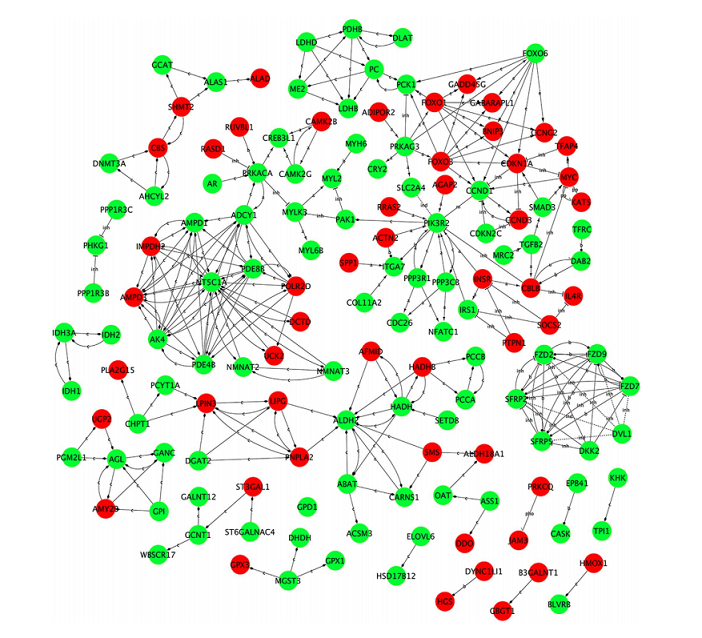
基因互作网络
Sun L et al,Sci Rep. 2016
注:红色圆点表示上调mRNAs,绿色圆点表示下调mRNAs。
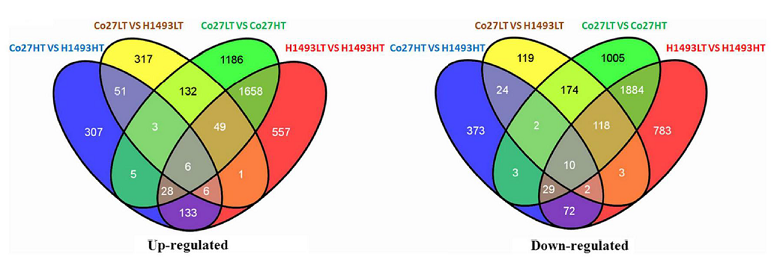
维恩分析
Chen et al., BMC Genomics, 2014
注:该图表示上调基因(左)和下调基因(下)的韦恩分析图,数字分别代表处于不同交集内的基因个数。
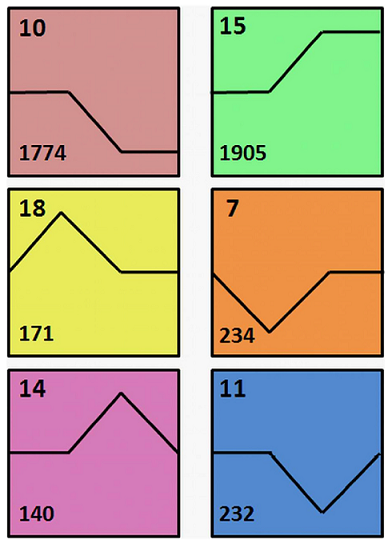
趋势分析
Chen et al., BMC Genomics, 2014
注:研究者基于趋势分析的众多结果,归纳、整合,最终锁定了几类变化趋势类型,进而更有针对性的开展后续工作。该研究中最终归纳出了6种显著性趋势,研究者选择了基因个数最多的两种趋势,对这些基因进行GO等深入分析。
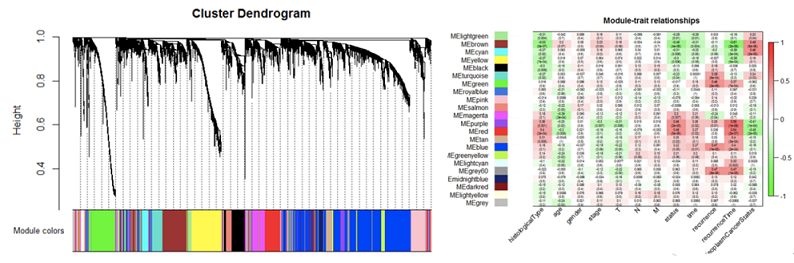
WGCNA分析 Wan et al., Exp Eye Res. 2018
注:左图表示基因聚类和模块鉴定的对应关系,高度共表达的基因群在聚类中处于相似分枝中;右图表示模块和表型相关性热图结果,方框内上面的数字是模块ME和表型数据相关性,下面括号内的数字为相关性的P值。
[1] Alteration of tumor suppressor BMP5 in sporadic colorectal cancer: a genomic and transcriptomic profiling based study. Molecular Cancer. 2018 Dec; IF=7.776
[2] Gefitinib for Epidermal Growth Factor Receptor Activated Osteoarthritis Subpopulation Treatment. EBioMedicine. 2018 Jun ;IF=6.183
[3] The GARP Complex Is Involved in Intracellular Cholesterol Transport via Targeting NPC2 to Lysosomes. Cell Rep. 2017 Jun; IF=8.032
[4] Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature. 2016 Feb; IF=41.577
[5] Hu, Y. et al. Interactions of OsMADS1 with floral homeotic genes in rice flower development. Mol. Plant 2015 Sep; IF=8.827
[6] Wang F, et al. Alternative splicing of the androgen receptor in polycystic ovary syndrome. Proc Natl Acad Sci U S A. 2015 Apr 14;112(15):4743-8. (IF=9.681)