服务热线02152235399
单细胞测序技术(Single Cell Sequencing)是在单个细胞水平上,揭示全基因组范围内的所有基因表达情况,包括鉴定组织细胞类型,反映不同样本间的细胞异质性和组织微环境。
【烈冰智造】于2021年9月发布了第一款用于单细胞测序的实验耗材——PanoCell®双通道微孔芯片;2022年9月推出了灵活度更高、兼容性更好的PanoCell®微孔芯片V2.0版。升级后的V2.0芯片将提供自研蜂巢板+BD平台原装进口单细胞分选试剂+华大T7超高通量测序仪的服务新方案!

1. 自主研发PanoCell微孔芯片,搭配BD Rhapsody单细胞平台+BD磁珠+BD全套原装试剂及protocol;



4.完善的质控流程和数据分析经验:烈冰自研的CytoNavigator 百万级通量数据分析系统确保数据的可靠性和质量,同时能够提供个性化、定制化的数据分析服务,帮助研究人员从海量的单细胞数据中获取有意义的信息。

样本类型:

组织、血液、培养的细胞系、制备好的单细胞悬液
注:若客户样本为组织,且无能力进行组织解离来获取单细胞悬液,烈冰将尽可能提供技术及实验上的帮助,但因不同类型样本的特异性,无法保证实验方法适用于所有类型组织。
质量要求:
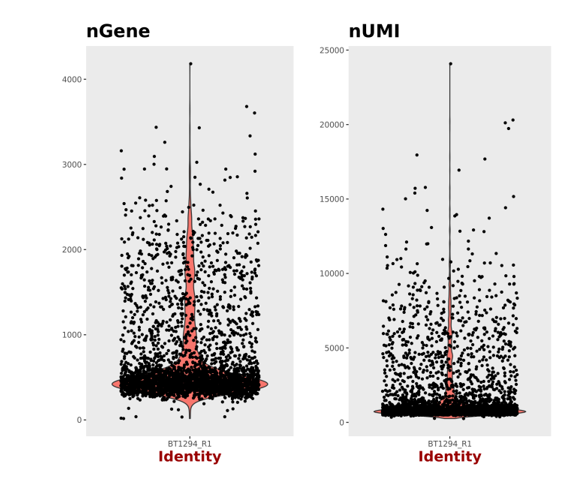
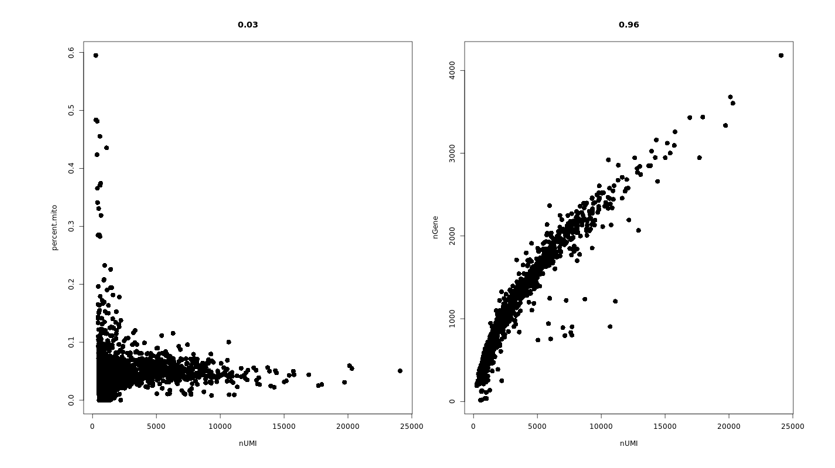
1. 细胞活性大于70%
2. 浓度为500-2000细胞/μl
3. 体积不小于200μl
4. 细胞培养基及缓冲液不能含Ca2+和Mg2+
5. 细胞体积小于40μm






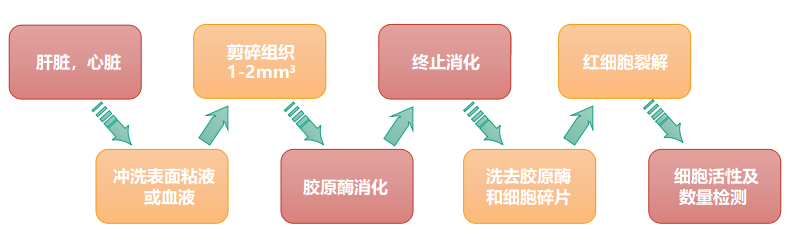
1. 单细胞悬液制备:根据样本特性选择合适的单细胞悬液制备方法,注意红细胞裂解;如若客户样本为已经制备好单细胞悬液,该步骤可以省略;
2. 细胞活性检测:细胞活性需大于70%;
3. 单细胞捕获:通过分选平台(BD)对每个细胞进行捕获;
4. 细胞/转录本标签添加:对结合磁珠标签的RNA进行逆转录引入CB和UMI;
5. 文库构建:对cDNA进行随机引物PCR扩增;
6. 上机测序:烈冰推荐Illumina Hiseq或NovaSeq测序平台,数据量100G/样本。

BD Rhapsody™单细胞分析系统的诞生基于 BD 在细胞生物学领域 40年的专业技术, 采用CytoSeq技术进行单细胞捕获。应用烈冰PanoCell微孔芯片(包含20W+的微孔(该数量级远大于Input细胞数量)),搭配BD平台磁珠+全套试剂+protocol,保证单孔中的单细胞捕获。同时避免了传统微流控系统中存在的概率碰撞影响捕获效率的问题,采用微孔捕获相对会有更好的捕获效率,保证Input细胞的全面使用。

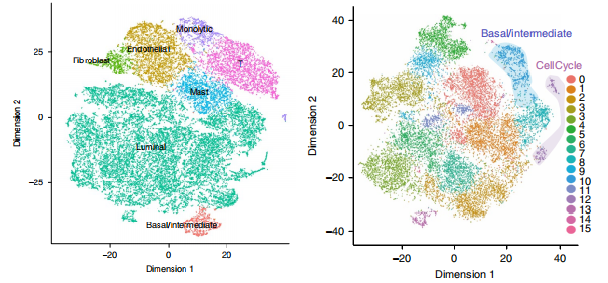
Chen, et al.Nature Cell Biology, 2021 Jan;23(1):87-98.
注:左图为前列腺肿瘤组织细胞亚群鉴定t-SNE图展示;右图细胞亚群分群情况
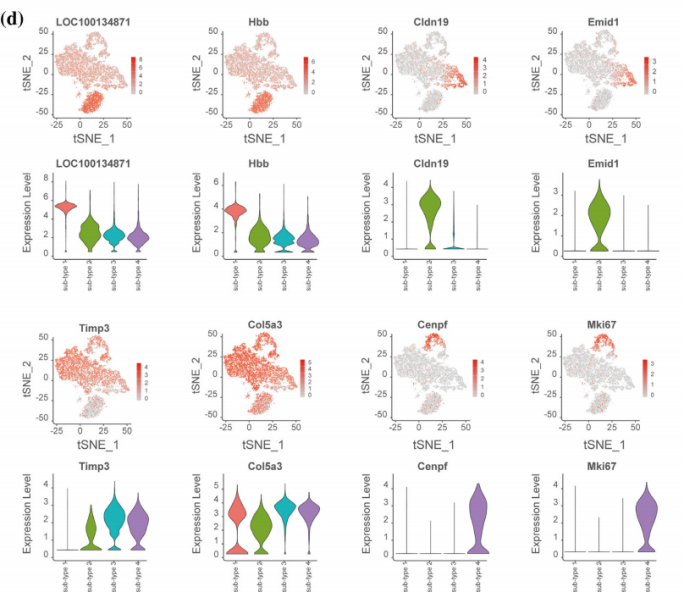
Zhang et al., Glia. 2021 Mar;69(3):765-778.
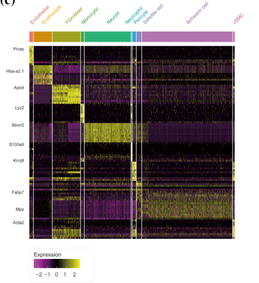
Zhang, et al. Glia. 2021 Mar;69(3):765-778.
注:该图为不同细胞亚群间差异基因聚类分析Heatmap图
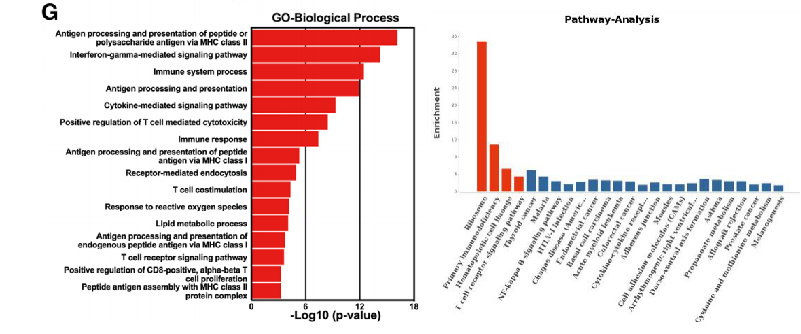
Zhang C, et al. J Immunother Cancer 2021;9:e002312.
注:该图为不同cluster之间差异基因显著富集的GO/Pathway条目
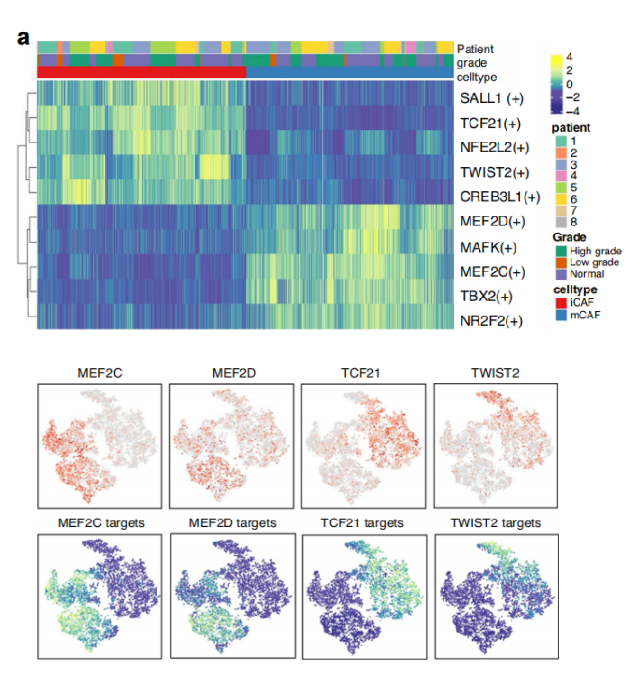
Chen, et al. Nature Communication 2020; 11:5077
注:该图为不同cluster之间转录因子调控强度heatmap图
Chen, et al.Nature Cell Biology, 2021 Jan;23(1):87-98.
注:该图为不同cluster之间信号同理调控强度热图
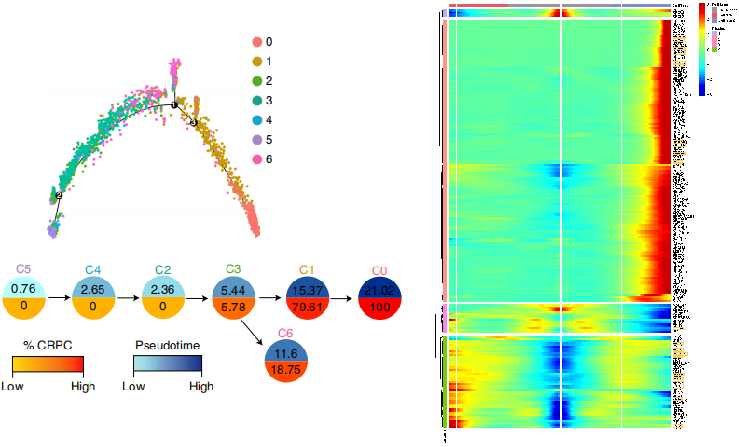
Chen, et al.Nature Cell Biology, 2021 Jan;23(1):87-98.
注:细胞间状态转换的pesudotime轨迹图(左)、轨迹中细胞占比饼图 和 Heatmap图(右)
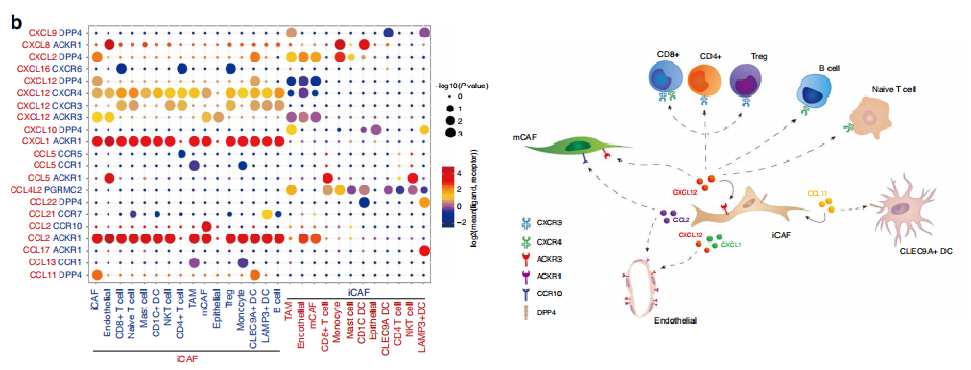
Chen, et al. Nature Communication 2020; 11:5077
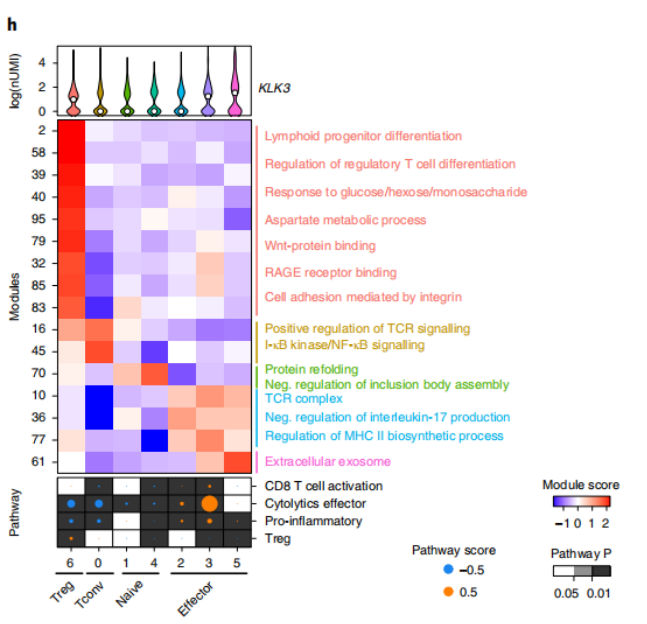
注:左图:横坐标表示细胞类型,纵坐标表示细胞间通讯配受体关系,圆圈大小表示显著性差异水平,圆圈颜色越红表示细胞间通讯关系越强
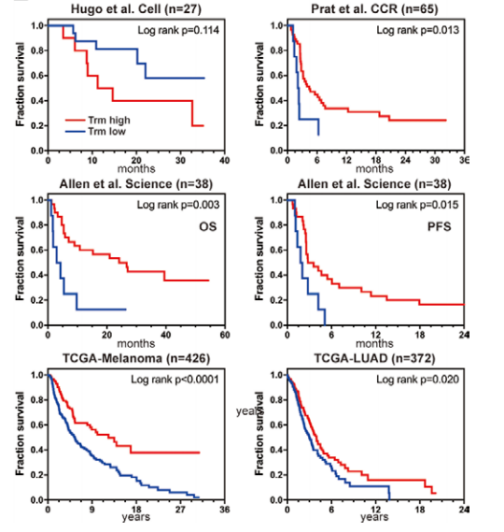
Zhang C, et al. J Immunother Cancer 2021;9:e002312.
[1] CD_99 G1 neutrophils modulate osteogenic differentiation of mesenchymal stem cells in the pathological process of ankylosing spondylitis.Ann Rheum Dis.2023 Oct;IF=27.4
[2] Immunosuppressive CD10+ALPL+ neutrophils promote resistance to anti-PD-1therapy in HCC by mediating irreversible exhaustion of T cells. Journal of Hepatology. 2023 Aug;IF=25.7
[3] Endothelial response to type I interferon contributes to vasculopathy and fibrosis and predicts disease progression of systemic sclerosis.Arthritis & Rheumatology. 2023 Jul; IF=13.3
[4] Schwann cells regulate tumor cells and cancer-associated fibroblasts in the pancreatic ductal adenocarcinoma microenvironment. Nature Communicationsl. 2023 Jul ; IF=16.6
[5] WT1+ glomerular parietal epithelial progenitors promote renal proximal tubule regeneration after severe acute kidney injury. 2023 Feb; IF=12.4
[6] Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. GUT. 2022 Jun; IF=24.5