服务热线02152235399
一 引言
1.1编写目的
进行该测试以及撰写此报告有以下几个目的
1.目前我们公司使用的几种经典算法,对于差异基因的筛选过于严格,导致结果后续分析的过程中遇到数据过少的情况。因此我们希望能够找到更好的筛选出更多可能性,却又不影响后续分析的参数来改善这种情况。
2.深入分析各种软件的各项参数。
3.深入分析各个软件对应各种数据的情况。
1.2背景
差异基因(diffGene)筛选的定义是用统计学的方法对高通量测序的结果进行筛选,跳出样本间有显著差异的基因。通过分析实验组,控制组;不同时间下的各个组之间的差异基因,可以帮助科学家找到与现象相关的基因,用于后续的实验验证。通过这种方法,大大加速了实验研究的进度。能够更加高效的进行生物学研究。
目前在差异基因筛选中,世界上最常用的软件有:DESeq,EBSeq,DEGSeq,以及其他的一些方法,比如TTest。目前的这些软件中,通用的优化方法有FPKM(RKPM),UpQuantile,median,TMM,TPM。在上述提到的这些软件中,分别使用到了这些方法中的一种或者几种方法,我们在讨论中会详细的解释这些结果。
鉴于我们目前对于差异基因的筛选结果过于严格,这样使得我们后续的分析不能很好的展开,所以开展这个分析项目,以改善目前的这种情况。
1.3用户群
主要读者:公司研发部,公司管理人员。
其他读者:项目及销售相关人员。
1.4 数据对象:
|
实验数据 |
实验数据 |
实验数据 |
|
客户潘洁雪所测的关于人类的2个样本,各3次重复的数据 |
程世平关于杨树的42个样的数据,各个数据有3次重复 |
周勇关于mouse的两个样本,各三次重复 |
1.5 测试阶段
本次测试一共分两个部分:
1. 各个软件参数摸索;
2. 调整以后参数对后续结果的影响分析。
1.6测试工具
R Version 3.0.0
EdgeR version 3.4.0
DEGSeq version 1.2.2
DESeq version 1.14.0
DESeq2 version 1.2.5
EBSeq version 1.3.1
1.7 参考资料
edgeR: dierential expression analysisof digital gene expression data User's Guide
Package ‘edgeR’
Dierential expression of RNA-Seq data at the gene level --the DESeq package
Package ‘DESeq’
How to use the DEGseq Package
DEGseq
EBSeq: An R package for differential expression analysis using RNA-seq data
Package ‘EBSeq’
二 测试概要
主要测试内容如下:
1. EBSeq参数测试
2. DEGSeq参数测试
3. DESeq参数测试
4. edgeR参数测试
5. 后续GO_Pathway分析验证
2.1工作计划进展
|
测试内容 |
计划开始时间 |
实际开始时间 |
计划完成时间 |
实际完成时间 |
工作完成情况 |
|
EBSeq参数测试 |
2013年10月25日 |
2013年10月25日 |
2013年10月27日 |
2013年10月27日 |
顺利 |
|
DEGseq参数测试 |
2013年10月28日 |
2013年10月28日 |
2013年10月29日 |
2013年10月29日 |
顺利 |
|
DESeq参数测试 |
2013年11月2日 |
2013年11月2日 |
2013年11月3日 |
2013年11月3日 |
顺利 |
|
EdgeR参数测试 |
2013年11月4日 |
2013年11月4日 |
2013年11月5日 |
2013年11月5日 |
顺利 |
|
GO_Pathway分析 |
2013年11月5日 |
2013年11月5日 |
2013年11月6日 |
2013年11月6日 |
顺利 |
|
撰写报告 |
2013年11月6日 |
2013年11月6日 |
|
|
顺利 |
表1.工作进度表
2.2测试执行
此次测试严格按照项目计划和测试计划执行,按时完成了测试计划规定的测试对象的测试。针对测试计划制定规定的测试策略,依据测试计划和测试用例,将网络数据以及我们观测的关键参数进行了完整的测试。
2.3测试用例
2.3.1功能性
1.测试如何提高差异基因筛选的方法与参数
2.测试提升灵敏度以后的参数对于后续分析的影响。
三 测试环境
3.1软硬件环境
|
硬件环境 |
服务器 |
|
硬件配置 |
CPU:Intel Xeon 2.66GHz *20 Memory:90GB HD:29TB |
|
软件配置 |
OS:Fedora release 14,Ubuntu 12.10 |
|
网络环境 |
100M LAN |
表2 软硬件环境
四 测试结果
4.1 使用默认参数进行测试
在这次的测试中,我们使用的默认脚本都是我们公司自己平台的脚本,并且也参阅了各个软件对应的脚本,确认了我们使用的脚本是默认脚本。在选择数据方面,我们选择了客户潘雪洁的关于人类的数据,这个数据包含了两个样本,每个样本具有3次重复。在使用各个软件进行处理以后,我们将得出的结果按照总共6个样本总和小于18,小于60,小于600的数据全部去除用以后续分析,进行这一步的主要目的是为了降低那些低丰度的表达量对于我们结果的影响。
|
使用包 |
筛选结果 |
|
DEGSeq |
90 |
|
DESeq |
12 |
|
EBSeq |
113 |
|
EdgeR |
8 |
*DEGSeq使用FPKM,其余均使用counts
表3 默认参数筛选出的差异基因数目
4.2参数优化探索
针对不同的软件,我们参照其用户手册以及函数说明进行了优化,在优化的过程中,我们着重考量了不同程序的预处理过程以及处理数据的核心部分的参数控制。针对我们优化以后的结果,我们将在讨论部分进行详细介绍。
|
使用包 |
筛选结果 |
|
DEGSeq |
90 |
|
DESeq |
697 |
|
EBSeq |
317 |
|
EdgeR |
8 |
*DEGSeq使用FPKM,其余均使用counts
表4 优化以后筛选出的差异基因数目
在上述结果中,我们看到只有DESeq以及EBSeq的结果有了显著提升,而其他两个软件的结果并没有变化。对于DEGseq,由于使用的是FPKM的值,我们在查找参数变化的时候并没有找到很好的参数,所以并没有产生变化。对于EdgeR,我们在测试的过程中,只发现了调节dipersion这个参数能够很好的改变结果,但是在查询这个参数的统计学意义的过程中,我们发现这个参数是用于反应样本之间的数值随机变化的显著性,即当这个参数为设置为0的时候就会认为数值之间的任何数值差异(即便相差1%)都会认为这两者在这次统计上是有差异的,于是我们参照了函数声明中求这个参数的函数进行估计,并没有发现软件支持的很好提高灵敏度的方法,同时我们认为人为的定义一个值在科学的意义上而言显得很不严谨。所以我们的放弃了这个软件的提升灵敏度的尝试。
4.3 DESeq默认参数与优化参数比较结果
在实验过程中,我们发现优化前后,DESeq的结果变化选出的基因从12个提升到了697个,提升了685个。是所有变化中最多的,为了确定结果对后续分析没有影响,我们使用输出的结果进行了后续的GO,Pathway分析。在处理之前,为了降低样本低丰度结果导致的影响,我们将输入结果按照六列样本数据和小于18,60,600的量分为了三次重复。我们希望通过这个做法来知道低丰度对于结果的影响。以下是我们详细分析的删除小于18的结果的GO,Pathway分析的结果。
使用默认参数
下调GO的结果,一共有103个。前30个结果截图如下
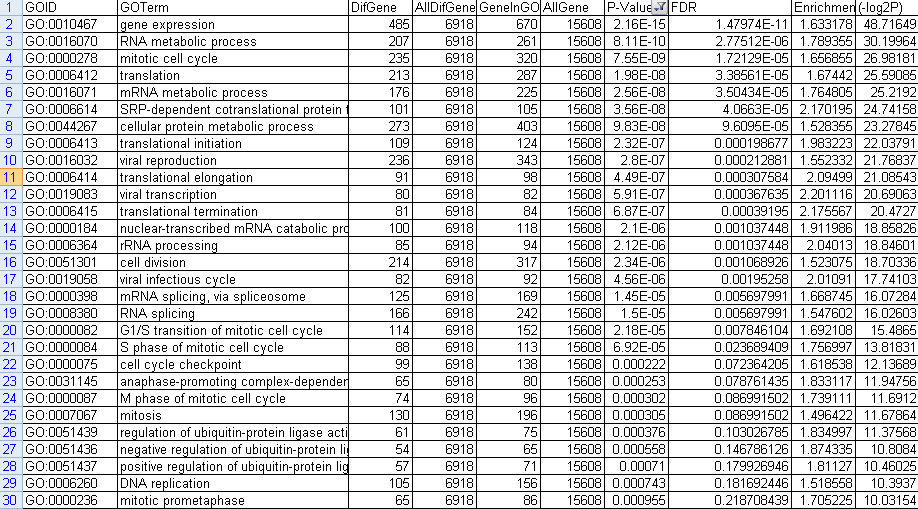
图1 下调GO筛选P-value结果图
上调GO的结果,一共有48个。前30个截图如下:
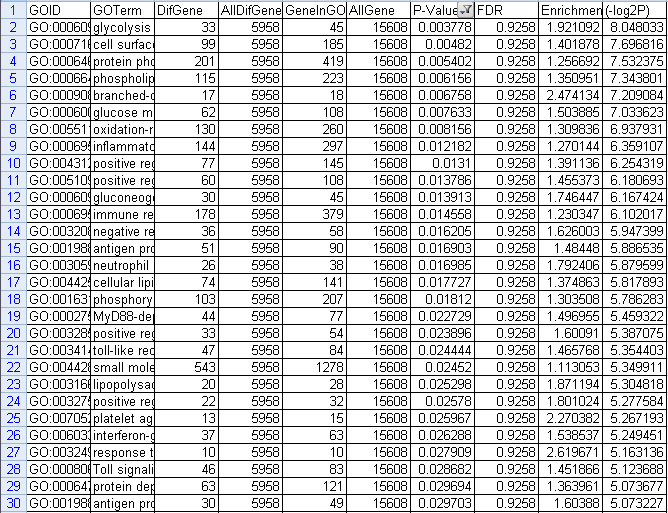
图2 上调GO筛选P-value结果图
按照FDR值小于0.05进行筛选,得出结果如下:
使用默认参数
下调GO的结果,一共20个,截图如下
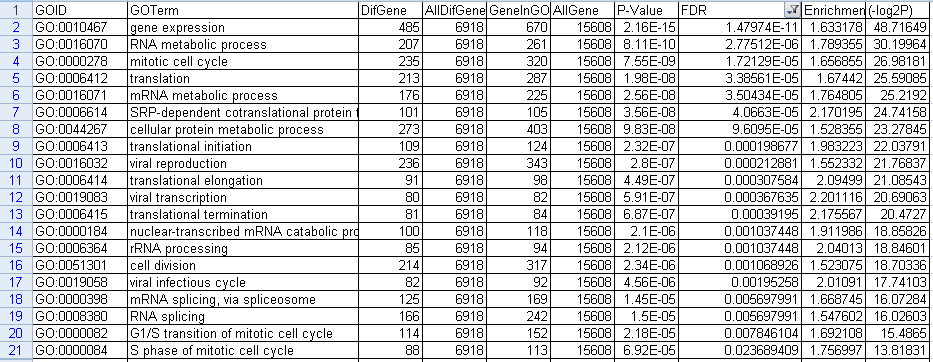
图3 下调GO筛选fdr结果图
上调GO的结果,
没有找到结果;前30个结果的截图如下:
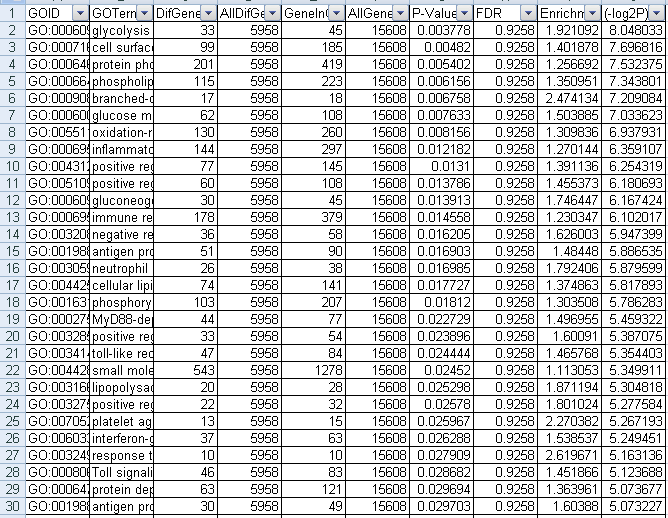
图4 上调GO筛选fdr结果图
之后我们使用我们优化的结果进行GO分析,其结果如下
下调GO的结果,一共有103个。前30个结果截图如下
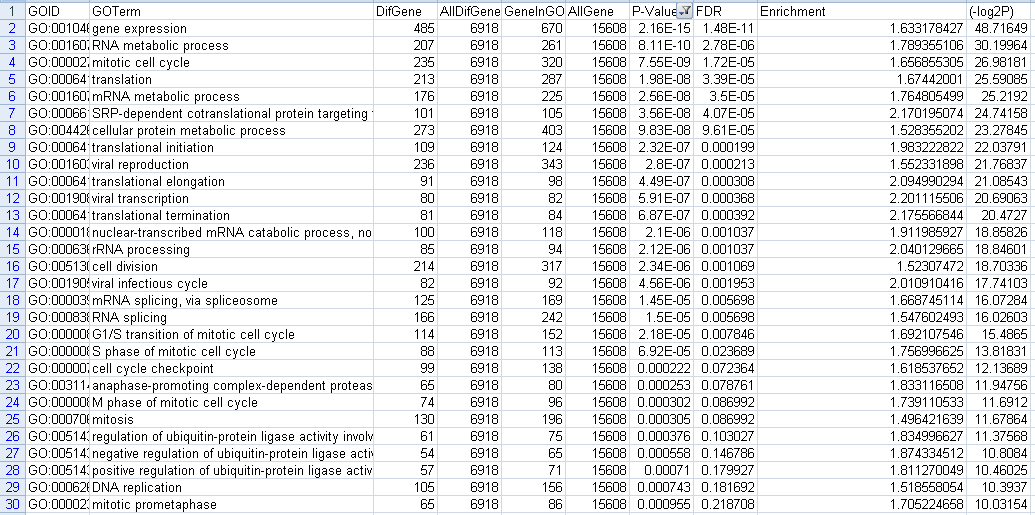
图5 下调GO筛选P-value结果图
上调GO的结果,一共有48个。前30个截图如下
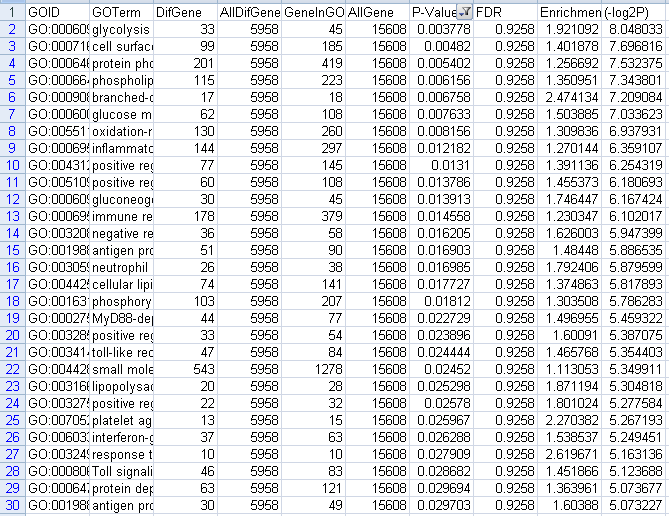
图6 上调GO筛选P-value结果图
根据fdr值小于0.05进行筛选
下调GO的结果,一共20个,截图如下
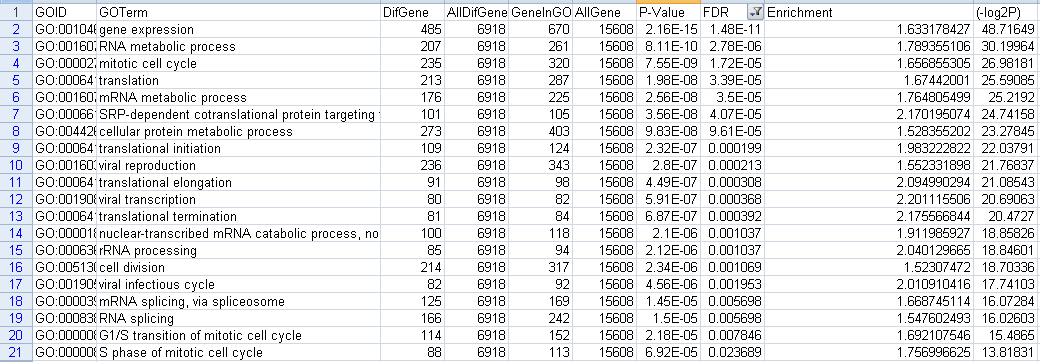
图7 下调GO筛选fdr结果图
上调GO的结果,
没有找到结果;前30个结果的截图如下:
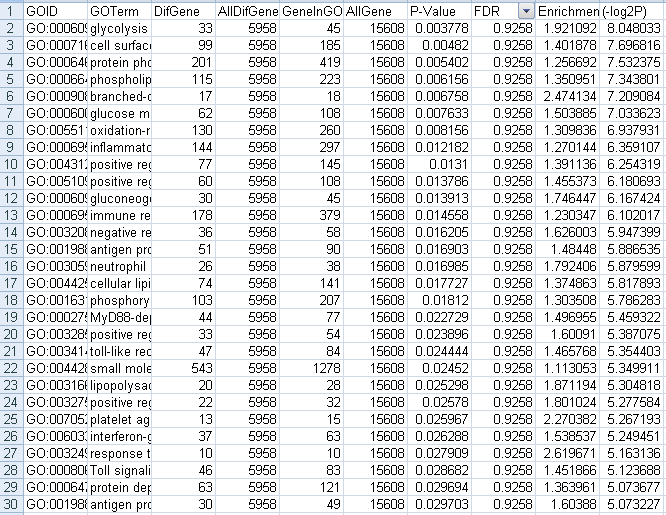
图8 上调GO筛选fdr结果图
我们将上述的结果进行了对照分析,发现两者的结果是一一对应的。这证明了我们提升灵敏度的方法对于GO分析没有影响。
接下来我们分析了Pathway的结果
使用默认参数:按照P-value小于0.05进行筛选的结果如下
下调代谢通路:一共筛选出19个结果,截图如下
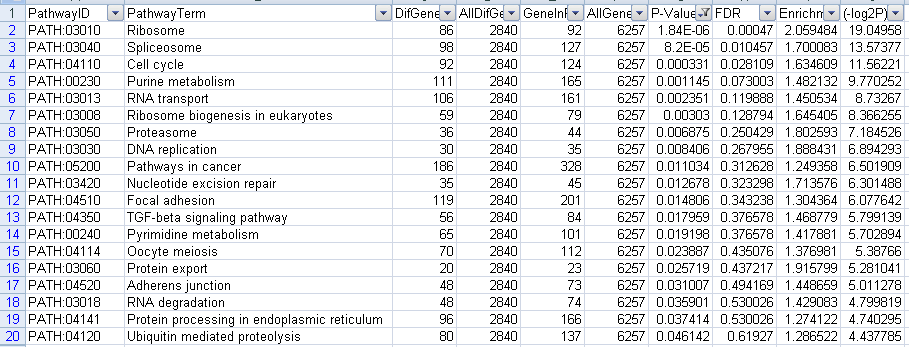
图9 下调Pathway筛选P-value结果图
上调代谢通路:一共筛选出50个结果,前30个截图如下:
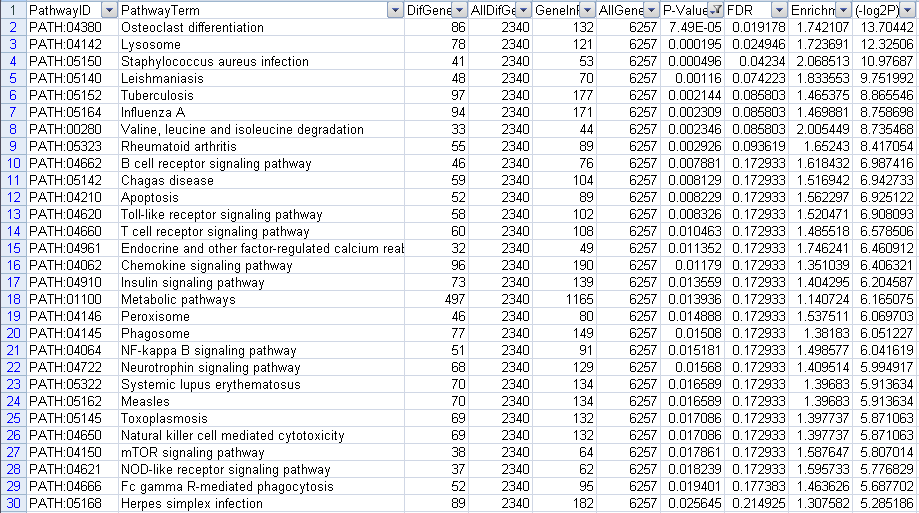
图10 上调Pathway筛选P-value结果图
使用默认参数:按照fdr小于0.05进行筛选,结果如下
下调代谢通路:一共筛选出3果,截图如下
![]()
图11 下调Pathway筛选fdr结果图
上调代谢通路:一共筛选出3结果,截图如下:
![]()
图12上调Pathway筛选fdr结果图
使用优化以后的参数
按照P-value小于0.05进行筛选的结果如下
下调代谢通路:一共筛选出19个结果,截图如下
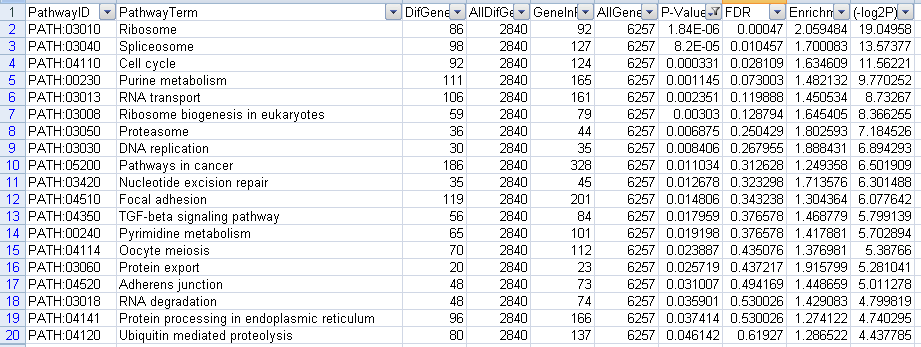
图13下调Pathway筛选P-value结果图
上调代谢通路:一共筛选出50个结果,前30个截图如下:
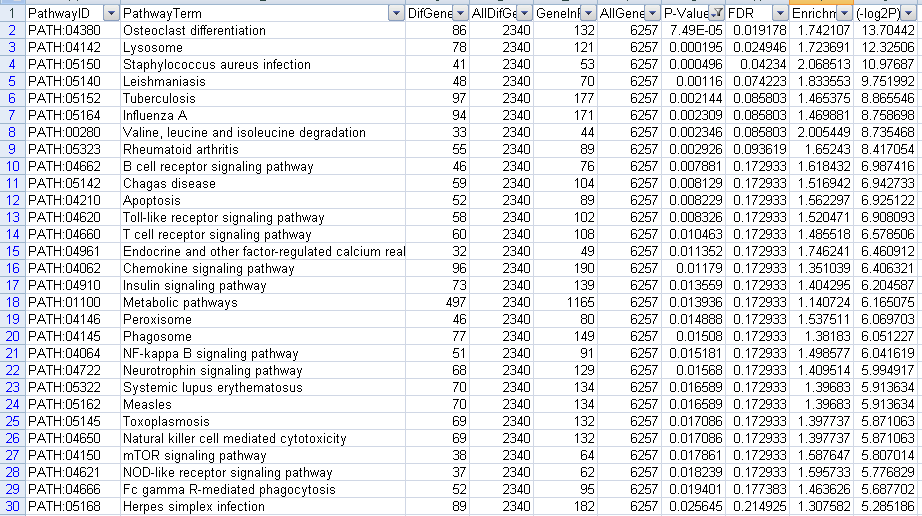
图14上调Pathway筛选P-value结果图
使用优化过后的参数:按照fdr小于0.05进行筛选,结果如下
下调代谢通路:一共筛选出3果,截图如下
![]()
图15下调Pathway筛选fdr结果图
上调代谢通路:一共筛选出3结果,截图如下
![]()
图16上调Pathway筛选fdr结果图
我们将上述的结果进行了对照分析,发现两者的结果是一一对应的。我们发现,无论是否按照排序还是飞排序的情况,我们的各种GO/PathwayID都是一一对应的。这证明了我们提升灵敏度的方法对于GO分析没有影响的预期。
这个结果证明我们对于DESeq使用的优化方法是可行的。
4.4 EBSeq默认参数与优化参数比较结果
在实验过程中,我们发现优化前后,EBSeq的结果变化选出的基因从12个提升到了697个,提升了685个。是所有变化中最多的,为了确定结果对后续分析没有影响,我们使用输出的结果进行了后续的GO,Pathway分析。与DESeq中的处理方式相似,在处理之前,为了降低样本低丰度结果导致的影响,我们将输入结果按照六列样本数据和小于18,60,600的量分为了三次重复。我们希望通过这个做法来知道低丰度对于结果的影响。以下是我们详细分析的删除小于18的结果的GO,Pathway分析的结果。
可以看出,在EBSeq中,筛选出的基因从113个提升到了317个,提升了204个。
之后我们做了GO分析,分析结果如下
筛选P-value小于0.05的值如下
使用默认参数
筛选下调GO结果,一共筛选出111个数据。前30个数据截图如下:
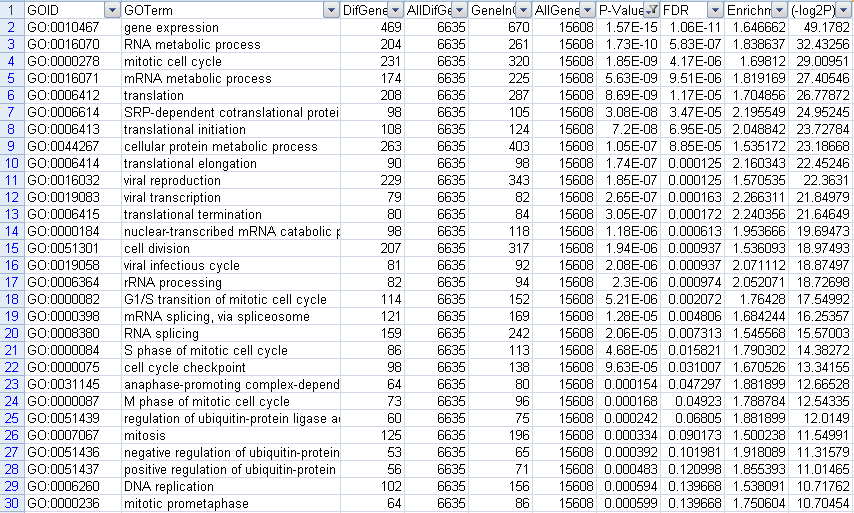
图16下调GO筛选P-value结果图
筛选上调结果,一共筛选出47个数据。前30个数据截图如下
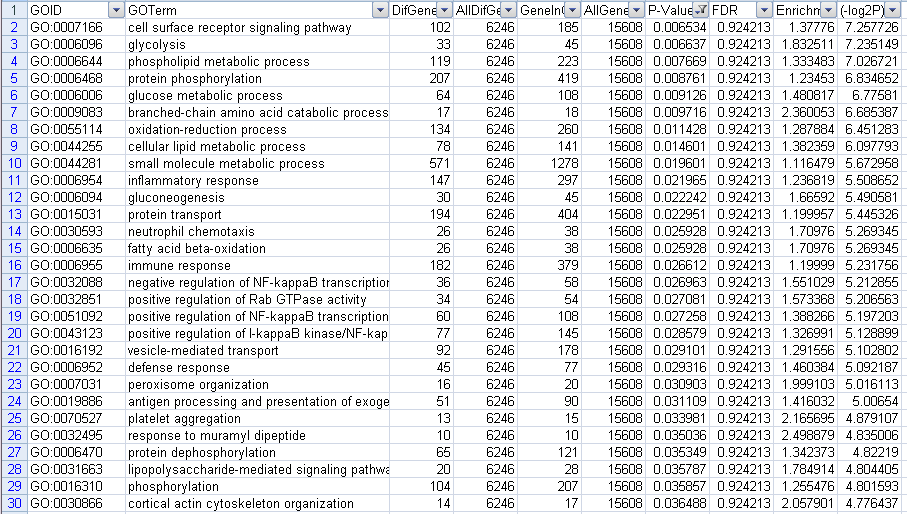
图17上调GO筛选P-value结果图
筛选fdr小于0.05的结果如下
筛选下调GO结果,一共得到23个结果,截图如下
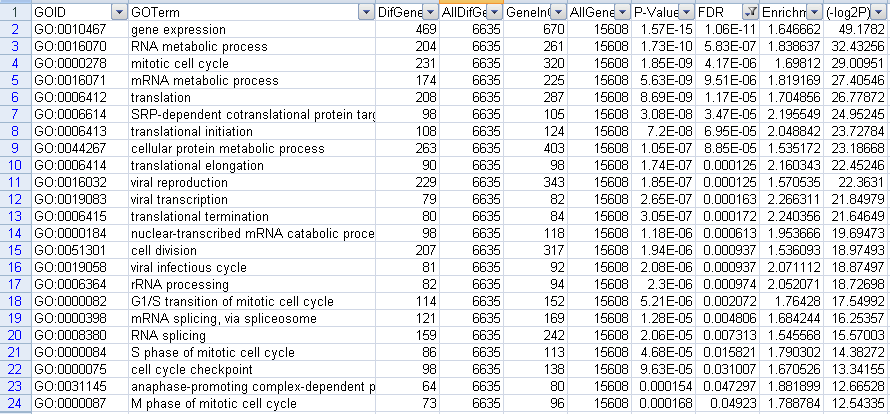
图18下调GO筛选fdr结果图
筛选上调GO结果,没有筛选到结果,截取前30个结果如下
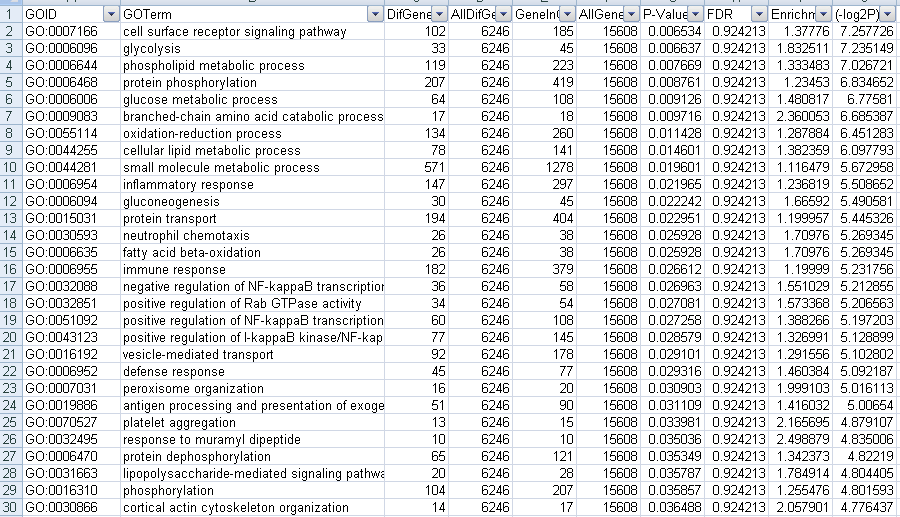
图19上调GO筛选fdr结果图
对于优化以后的参数
我们筛选p-value小于0.05
筛选下调GO结果,一共得到111个结果,前30个结果如下
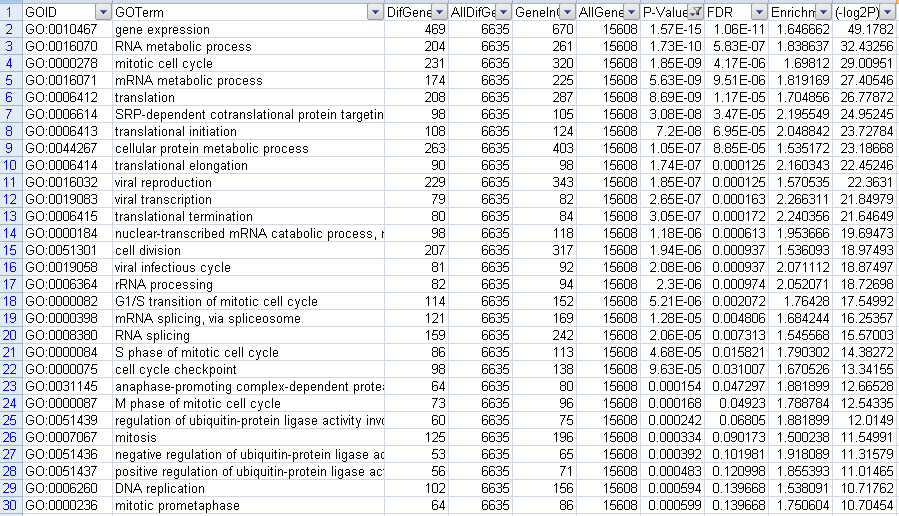
图20下调GO筛选P-value结果图
筛选上调GO结果,一共得到47个结果,前30个结果如下
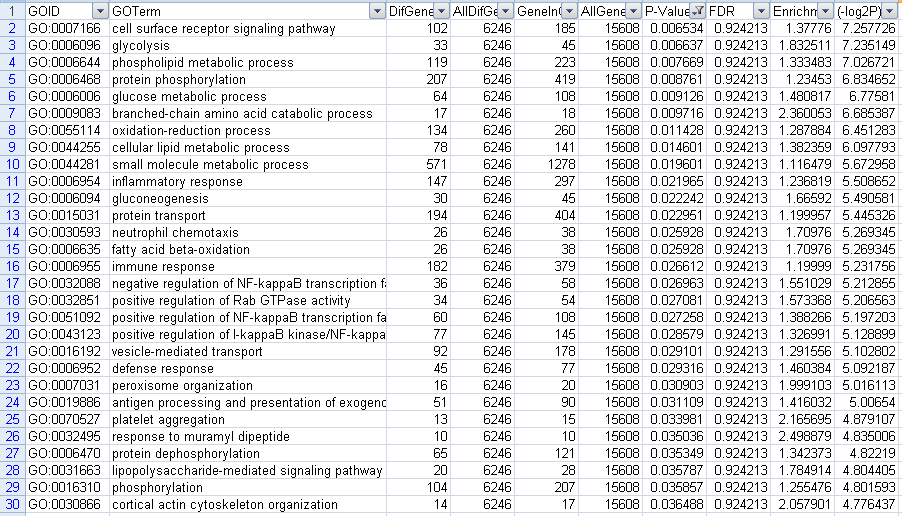
图21上调GO筛选P-value结果图
接下来我们分析了Pathway的结果
使用默认参数
按照P-value小于0.05进行筛选的结果如下
下调代谢通路,一共筛选出19个结果,截图如下
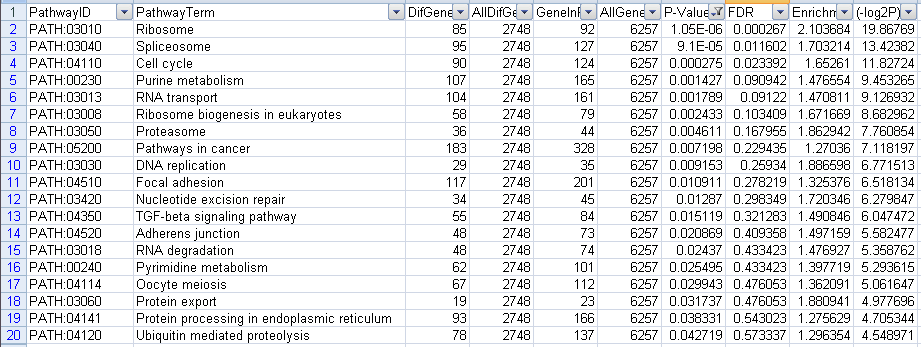
图22下调GO筛选P-value结果图
上调代谢通路,一共筛选出43个结果,前30个结果截图如下
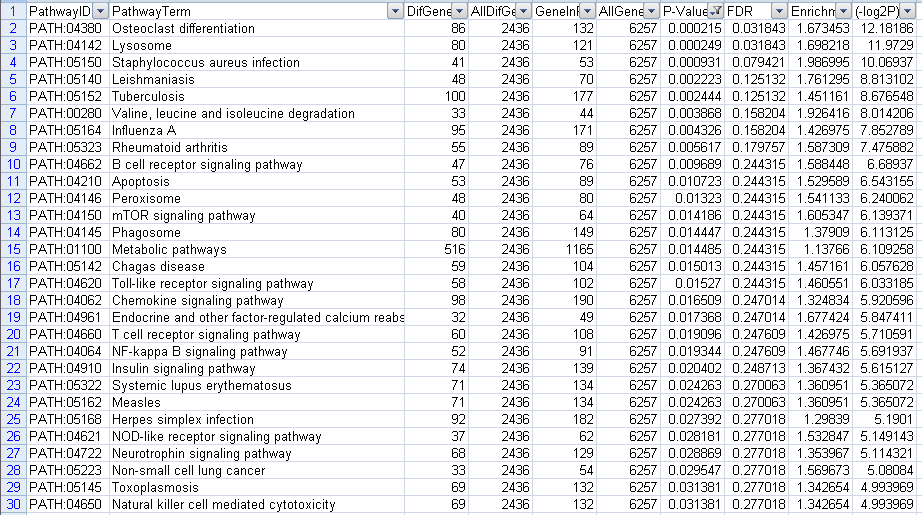
图23上调GO筛选P-value结果图
我们使用fdr小于0.05进行筛选
下调代谢通路,一共筛选出3个结果,截图如下
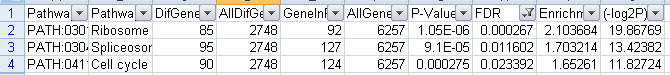
图24下调GO筛选fdr结果图
对于上调通路,一共筛选出2个结果,截图如下:
![]()
图25上调GO筛选fdr结果图
对于优化以后的参数
我们筛选p-value小于0.05
筛选下调GO结果,一共得到19个结果,截图如下
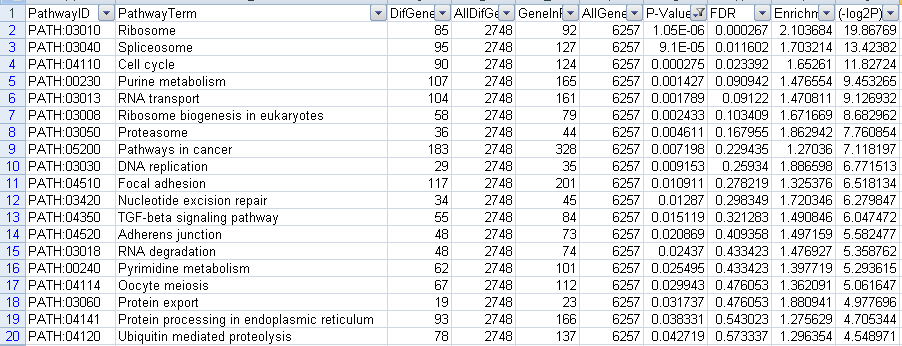
图26下调GO筛选P-value结果图
筛选上调GO结果,一共得到43个结果,前30个结果截图如下
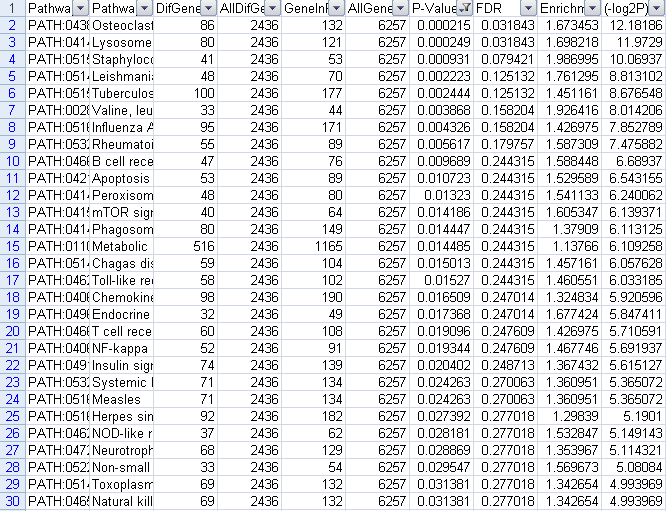
图27上调GO筛选fdr结果图
筛选fdr小于0.05的结果如下
筛选下调GO结果,一共得到3个结果,截图如下
![]()
图28下GO筛选fdr结果图
筛选上调GO的结果,一共得到2个结果,截图如下
![]()
图29上调GO筛选fdr结果图
我们比较使用默认参数与优化以后的参数的变化,发现这两者做出的GO,以及Pathway的结果是一致的。无论是否按照排序都是出现相同的结果。
所以我们可以知道,我们对于EBSeq的处理方法是可行的。
4.5 EdgeR等其他软件的测试
正如上述所说,我们在测试edgeR以及DEGSeq的过程中,发现了一些能够改进的方法, 但是由于科学必须遵守严谨的作风。所以我们并没有盲目的采用这些做法。
五.测试结论与讨论
5.1 DESeq参数改进及解释
对DESeq的方法而言,程序默认的R脚本如下:
|
library(DESeq2) data = read.table(fileName, he=T, sep="\t", row.names=1)
colData=data.frame(conds)
cds = DESeqDataSetFromMatrix( data, colData,,formula(~ conds) ) cds = estimateSizeFactors(cds) cds = estimateDispersions(cds)
res = nbinomTest( cds, "p", "c" ) write.table( res, file="/home/novelbio/hxl_temp/DifferenceExpression_result/CPVS_DESeq.xls",sep="\t",row.names=F ) |
我们优化的脚本如下
|
filePath = "/home/novelbio/hxl_temp" fileName = "/home/novelbio/hxl_temp/All_Counts600.txt" setwd(filePath) library(DESeq) data = read.table(fileName, he=T, sep="\t", row.names=1)
conds = factor( c("c", "c", "p", "p", "p", "c") ) cds = newCountDataSet( data, conds ) cds = estimateSizeFactors(cds) cds = estimateDispersions(cds,method="per-condition",sharingMode="gene-est-only")
res = nbinomTest( cds, "p", "c" ) write.table( res, file="/home/novelbio/hxl_temp/CPVS_DESeq600_optimization.xls",sep="\t",row.names=F ) |
在上述命令中,我们的改动使用的红色进行标注。其解释如下:estimateDispersions这个函数的功能是用于估计dispersion的参数,正如我们上述所说,这个参数是用于衡量样本之间数据变化对于很衡量样本差异的灵敏度的值,当这个值设为0的时候,就意味着即使两个样本有一点点的差异,也认为这个数据是能够反映基因表达的差异的,也就是说,这个数据越小,越能说明灵敏的反映基因差异的显著性。在这里,我们额外的加入了两个参数,分别是method,这个参数是指定计算dispersion的方法,而我们指定的per-condition是指,针对不同的样本重复计算出不等的dispersion值,在我们的例子中,我们一个样本有三组重复,就意味着每一次重复都对应了一个离差值。而通过查阅函数手册,我们知道计算离差的方法一共有4种,分别是pooled,pooled-CR,per-condition,blind。其中,第一种pooled的方法最为粗糙, 这个方法对于所有的数据而言,只估计了一个经验性的dispersion值,并且将这个值用于所有的样本,显然这个做法并不严谨,在使用的过程中我们并没有使用这种做法。第二种pooled-CR的做法是通过Cox-Reid adjusted profile likelihood的方法估计出dispersion值,显然这种做法的运算速度要慢于第一种做法。第三种方法就是我们采用的方法,,对于不同条件的数据分别计算不同的dispersion值。但是这种方法在用于多种条件分析的情况(并非样本重复)时,并不适用。即当我们在同一次运行中,不应该使用多个比较(比如同时分析时间点以及实验,对照组,但是分开是可以使用的)。第四种方法blind的做法是认为所有样本都是同一条件下的不同重复,忽略样本的标记(labels),并计算经验性的dispersion值。即便是没有生物学重复的情况下也能使用,但是这种方法会导致样本失去某些重要的东西(loss of power),显然这种方法也是不严谨的。综合考虑这些方法的原理以及我们在实验中的结果,我们认为使用per-condition的方法显然是更加合理的。我们加入的另一个参数sharing-mode的作用是选择将优化以后的数据还是没有优化的数据进行后续的分析,在这里,一共有三个参数,fit-only,maximum,gene-est-only。这三个参数的解释如下:fit-only的作用是只使用优化以后的数据。函数手册中建议只有当具有少量的重复的时候才能使用这种方法。Maximum的方法是在优化的数据以及未优化的数据中选取较大值。函数手册建议当只有3个重复以下的时候使用这个参数。而第三个参数,gene-est-only在样本重复比较多并且dispersion值是可靠的时候适用,当样本重复少的时候,这个参数会比较灵敏的反映出样本变化情况。我们在测试的过程中发现,当sharing-mode选择gene-est-only的时候,得出的结果能够很好的提高灵敏度,这与我们进行试验的目的是一致的。所以我们选择了这个参数。而method选择的目的则是为了更好的提高我们实验过程中的严谨性。
通过后续的GO,Pathway分析,我们发现我们优化以后以及优化以前的的数据做出的GO,Pathway分析的结果是一致的,这直接的证明了我们处理方式是合理的。我们可以看出,无论是使用P-value还是FDR进行筛选,其结果都是一致的。由于GO,Pathway分析后续的步骤都是相同,即,使用相同的数据其结果也是相同的,所以我们并没有做画图分析。
5.2 DESeq脚本中其余几个函数的参数尝试
总结DESeq中的几个参数,在estimateDispersions函数中还有一个我们测试过的参数,fitType,这个参数的作用是设置用于计算dispersion-mean relation关系的方法,一共有两个参数,parametric,以及local,前者是使用公式dispersion 与asymptDisp, extraPois之间的公式进行计算,而local则是指使用locfit包来进行计算。我们在实验的过程中,发现这两个值的选取对于结果并没有影响,所以我们并没有把这个参数的计算放入结果的讨论中。
estimateSizeFactors,在DESeq设置sizeFactor的方法中,默认使用median 的方法。官方手册上所说在低count数的情况下使用shorth的方法我们并没有在R语言中找到。指定方法所使用的参数是locfunc。
nbinomTest,是整个函数中计算差异结果的核心。我们查阅了函数手册,这个函数并没有能够改变灵敏度的参数。我们在实验的过程中也没找到对应的参数,所以并没有进行优化。
5.3 EBSeq参数改进及解释
程序默认的R脚本如下:
|
filePath = "/home/novelbio/soft/NBCnew6/rscript/tmp/"
fileName = "/home/novelbio/hxl_temp/All_Counts18.txt" setwd(filePath) library(EBSeq) data = read.table(fileName, he=T, sep="\t", row.names=1) data = as.matrix(data) size = QuantileNorm(data, 0.75)
dataRun=data[,c(1, 2, 3, 4, 5, 6)] sizeRun = size[c(1, 2, 3, 4, 5, 6)] EBOutRun<- EBTest(Data=dataRun, Conditions=as.factor(c("c", "c", "p", "p", "p", "c")),sizeFactors=sizeRun, maxround=5) GenePP1=GetPPMat(EBOutRun) FDR<-GenePP1[,1]
c<- unlist(EBOutRun$C1Mean) p<- unlist(EBOutRun$C2Mean) LogFC=log2(PostFC(EBOutRun)$RealFC)
out<-cbind(c,p, LogFC,FDR ) colnames(out)<-c("c","p","LogFC", "FDR") write.table(out, file="/home/novelbio/hxl_temp/CPVS_EBSeq18_withOutOptimization.xls", row.names=TRUE,col.names=TRUE,sep="\t") |
我们优化以后的参数如下:
|
fileName = "/home/novelbio/hxl_temp/All_Counts600.txt" library(EBSeq) data = read.table(fileName, he=T, sep="\t", row.names=1) data = as.matrix(data) size = QuantileNorm(data,0.75)
dataRun=data[,c(1,2,3,4,5,6)] sizeRun = size[c(1,2,3,4,5,6)] EBOutRun<- EBTest(Data=dataRun, Conditions=as.factor(c("C", "C", "P", "P", "P", "C")) ,sizeFactors=sizeRun, maxround=1 ,ApproxVal=0.3) GenePP1=GetPPMat(EBOutRun) FDR<-GenePP1[,1] |
|
C<- unlist(EBOutRun$C1Mean) P<- unlist(EBOutRun$C2Mean) LogFC=log2(PostFC(EBOutRun)$RealFC)
out<-cbind(C,P, LogFC,FDR ) colnames(out)<-c("DF","HF","LogFC", "FDR") write.table(out, file="/home/novelbio/hxl_temp/CVSP_EBSeq600_optimization.xls", row.names=TRUE,col.names=TRUE,sep="\t") |
在上述命令中,我们的改动使用的红色进行标注。其解释如下:maxround是指EBTest迭代的次数,迭代次数越多,得出的结果越少。程序默认的迭代次数是5次。而我们公司平台指定的次数是15次,这样虽然看似很严谨,但是也会过滤掉很多有用的信息,显然,这不是将实验结果最大化利用的最合理的方法。在实验的过程中,我们发现即便这个参数设置成为1,也不会对后续的分析产生影响。所以我们在这里就大胆的设置成为1次迭代。ApproxVal的英文解释是The variances of the transcripts with mean < var will be approximated as mean/(1-ApproxVal),是用于调整mean的值的一个参数,我们在实验的过程中发现,当这个值设置为0.3的时候,是最能提升差异基因筛选的结果的。而在后续的实验中,我们发现这个结果的变化并不会对结果产生影响,所以我们也接受了这个值的选择。
纵观整个EBSeq的脚本,函数手册及其用户帮助,我们并没有找到与DESeq,edgeR中都具有的参数dispersion,我们认为这可能是导致EBSeq结果提升不如DESeq明显的主要因素。
5.4EBSeq脚本中其余几个函数的参数尝试
在EBSeq中,运行的代码可以大致分为两个部分,数据预处理以及差异基因筛选,在数据预处理中,我们发现计算size这个变量的函数一共有如下几个:quantileNorm,这个函数是使用百分位点的方法进行数据标准化;RankNorm,这个函数是利用rank标准化方法进行标准化。另外一种标准化方法是MedianNorm,这个方法使用Anders et. al., 2010.提出的中位数标准化方法进行标准化。在我们测试的过程中,针对这一组数据,我们发现不同的标准化方法得出的结果是几乎一样的。针对具体方法的具体使用对象,我们并没有找到各自针对的情况。在函数EBTest中,还有几个参数,比如pool,函数手册建议在没有样本重复的时候设置这个参数为TRUE,而PoolLower,PoolUpper两个参数这是指定只用于分析的数据集,即只取在这两者范围内的数据集用于分析。其余的参数我们并没有发现能够改善分析步奏的严谨性或者提高差异基因筛选灵敏度能力的参数,所以并没有进行指定。对于这些参数,我们认为使用软件默认的参数就不错了。
5.5edgeR参数改进及解释
虽然在实验的过程中,我们并没有很合理的改善edgeR筛选差异基因的灵敏度的能力,但是我们还是对这个软件进行了探索。
|
filePath = "/home/novelbio/hxl_temp" fileName = "/home/novelbio/hxl_temp/diff.txt" setwd(filePath) data2 = read.table(fileName, he=T, sep="\t", row.names=1) library(edgeR) bcv = 0 plus = 0 data = read.table(fileName, he=T, sep="\t", row.names=1) data = data + plus group = factor( c("c", "c","p", "p", "p", "c" ) )
#######################一般方法#####################
Normfactors=calcNormFactors(data,method="RLE") p = DGEList(counts=data,group=group) p = estimateCommonDisp(p) p= estimateTagwiseDisp(p)
result = exactTest(p, pair=c("c","p" ) ,dispersion=p$tagwise.dispersion);
result = exactTest(p, pair=c("c","p" ) ); resultFinal=result$table; result2=cbind(resultFinal,fdr=p.adjust(resultFinal[,4])); write.table(result2, file="/home/novelbio/hxl_temp/CPVS_EdgeR.xls",sep="\t")
###############LRT_Method#################
design=model.matrix(~group) m = DGEList(counts=data,group=group) m=estimateGLMCommonDisp(m,design) m <- estimateGLMTrendedDisp(m,design) m<- estimateGLMTagwiseDisp(m,design) fit <- glmFit(m,design) lrt <- glmLRT(fit,coef=2)
lrtFinal=lrt$table; lrt2=cbind(lrtFinal,fdr=p.adjust(lrtFinal[,4])); write.table(lrt2, file="/home/novelbio/hxl_temp/CPVS_EdgeR.xls",sep="\t")
|
在EdgeR中,对于单组数据的程序如下:
|
filePath = "/home/novelbio/hxl_temp" fileName = "/home/novelbio/hxl_temp/diff.txt" setwd(filePath) data2 = read.table(fileName, he=T, sep="\t", row.names=1) library(edgeR)
bcv = 0 plus = 0 data = read.table(fileName, he=T, sep="\t", row.names=1) data = data + plus group = factor( c("c", "c","p", "p", "p", "c" ) )
#######################一般方法#####################
Normfactors=calcNormFactors(data,method="RLE") p = DGEList(counts=data,group=group) p = estimateCommonDisp(p)
result = exactTest(p, pair=c("c","p" ) ,dispersion=p$common.dispersion);
result = exactTest(p, pair=c("c","p" ) ); resultFinal=result$table; result2=cbind(resultFinal,fdr=p.adjust(resultFinal[,4])); write.table(result2, file="/home/novelbio/hxl_temp/CPVS_EdgeR.xls",sep="\t")
|
我们通过查询edgeR的函数手册,发现这个结果中也有关于dispersion的函数,通过查阅函数手册,发现有几种可以指定这个值的方法,最简单的也是我们现在使用的,是先指定一个bcv(这个值是dispersion的平方根),我们平台目前指定的是0.3,然后在exactTest函数中指定dispersion=bcv^2。在函数手册中,有几个关于这个值的经验数值,比如对于人类数据这个值去0.4,对于相似的模式生物(identical model organisms)可以取0.1,对于重复样本可以取0.01(technical replicates).在我们看来,使用这种方法并不利于批量化作业,而且从科学的角度而言也显得不严谨。其余估计dispersion的函数有estimateTagwiseDisp()这个函数与DESeq中的per-condition是类似的,都是按照列来估计dispersion。而estimateCommonDisp()这个函数则和DESeq中的pooled的方法类似,也是对于所有数据估计一个dispersion值,我们认为这样的做法并不严谨,因此也就没有采用。
在我们上述贴出来的代码中,一共分为3个部分,分别是计算多个样本重复的两种方法(存在检验(目前平台使用的方法)以及线性拟合实验(LRT))的方法,在我们的测试中,这两种方法对于结果没有明显影响。所以我们认为使用哪一种方法都是可行的。在用户手册中,作者建议在多个因素(maulti factor)的情况下使用LRT的方法。
在我们的测试过程中,我们发现,当bcv设置为0的时候,结果能够提升到1200个左右,因此,我们认为在满足条件(technical replicates)的时候,设置bcv=0.01的条件是可以提升这个结果的。
5.6 DESeq以及DESeq2
在测试DESeq的过程中,我们发现这个软件推出了新版本,DESeq2。查阅相关手册,我们发现DESeq2相比DESeq最大的改变是能够直接读取序列文件,然后自己计算counts数。其余并没有发现变化。查阅DESeq2的用户手册,我们发现如下变化:
1.使用SummarizedExperiment用于储存输入文件,即时计算结果,以及结果。
2. Maximum a posteriori estimation of GLM coecients incorporating a zero-mean normal prior withvariance estimated from data (equivalent to Tikhonov/ridge regularization). This adjustment haslittle eect on genes with high counts, yet it helps to moderate the otherwise large spread in log2fold changes for genes with low counts (e. g. single digits per condition). 3. Maximum a posteriori estimation of dispersion replaces the sharingMode options fit-only or maximum of the previous version of the package.
4.所有的估计都是基于通用的线性模型(generalized linear model),包括在两个条件的情况(之前的版本中使用的存在检验方法)
5.加入了Wald检验方法,同时之前的释然率检验的方法也同样能够使用。
6.允许输入标准化因子(normalized factors)
5.7 各个软件的输入文件
|
软件 |
输入数据 |
是否能够输入标准化数据 |
|
DESeq |
Counts |
否 |
|
DESeq2 |
Counts/ |
否 |
|
EBSeq |
Counts |
否 |
|
DEGSeq |
RPKM |
是 |
|
edgeR |
Counts |
否 |
表5 各个软件输入文本以及是否能够输入标准化数据
以上信息我们是通过查阅相关用户手册获得,并未在实验中进行验证。
5.8 DESeq涉及多个条件下的method选择
在实际生产中,我们发现,当涉及多个条件的情况是,DESeq中选择per-condition的方法并不能进行计算,因此我们必须选择这种情况下能够使用的方法。因此我们使用了实际生产的数据进行分析得出的结果如下:
|
方法 |
2nH-Fu |
2nH-Mu |
2nL-2nH |
2nL-2nMix |
2nL-Fu |
|
Pooled |
17 |
67 |
23 |
5 |
27 |
|
Pooled-CR |
27 |
67 |
31 |
44 |
44 |
|
Blind |
X |
X |
X |
X |
X |
|
Per-condition |
X |
X |
X |
X |
X |
|
默认 |
1 |
5 |
0 |
0 |
2 |
从上述结果可以看出,效果最好的是pooled-CR,而Blind的method对应也报错(与per-condition的一样),而pooled的结果和pooled-CR的结果都比默认参数的效果好,因此我们可以使用这两种方法,之后我们比较了对应两种条件下pooled和per-condition的效果,发现per-condition的效果(11903)比pooled-CR(8848)的效果要好。因此对应两种条件下的选择不变。
六.测试总结
1.在测试过程中,我们成功在不影响后续结果的情况下提升了差异基因灵敏度的方法,并且证明了我们方法的正确性
2.对于edgeR以及DESEq而言,能够很好的控制其灵敏度的参数是控制dispersion的值。对于EBSeq而言,则是控制迭代的次数以及控制ApproxVal的值。
3.由于edgeR中没有能够提供一个比较科学的获取dispersion的方法,所以我们认为并没有很好的改变结果。但是值得注意的一点是,在技术手册中提到,当重复是属于技术重复的时候,是可以使用bcv=0.01这个值的。