服务热线02152235399
一、find_circ简介
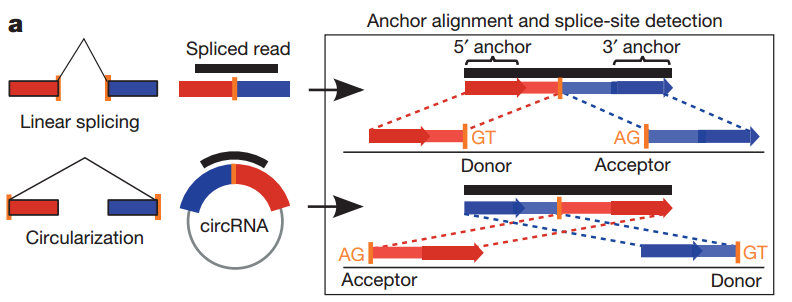
find_circ 通过识别junction reads 来预测circRNA和参考基因组比对完之后,首先剔除和基因组完全比对的reads,保留没比对上的reads, 这部分reads 直接比是比对不上基因组的,因为其来自不同的外显子区域,直接比对的话不允许这么大片段的缺失,那么如何区分剪切的spliced read 和 来自环状RNA的junction read呢,从上面的示意图我们可以直接看出,spliced read 的两部分比对在基因组上的前后位置和转录本中的位置保持一致,而来自circRNA的junction read 其比对的位置是相反的;具体操作的时候,首先从junction read的5'端和3'端取一部分序列,分别叫做5' anchor 和 3" anchor, 如果两个序列比对的位置是相反的,这条reads 就是一个可能的junction read, 然后将anchor read 一直延伸,直到连接处为止,如果到连接处为止序列都能够完全匹配,再看连接点处的剪切模式是否符合AG-GT的剪切模式,如果以上条件都满足,就认定这是一个circRNA。
通过对不同组织中的circRNA 进行预测,发现不同组织中存在的circRNA的数量和种类是有差别的,就是说circRNA 具有组织特异性。
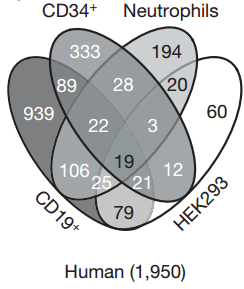
对人的不同的细胞系进行环状RNA的预测,发现不同细胞系中存在的circRNA的数量和种类都是有差异的,说明circRNA 具有组织特异性;
预测得到的circRNA如何验证:
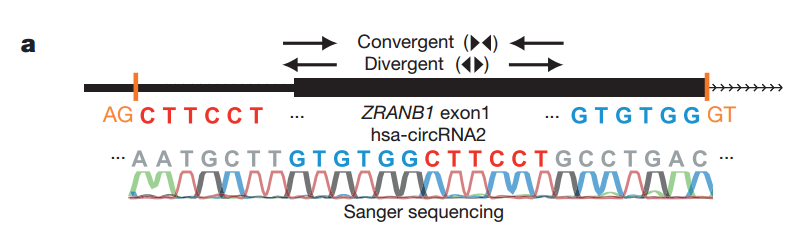
以上面的circRNA 为例,通常为扩增这一区域的线性RNA, 我们采用的引物都是Convertgent 所示的方向,但是为了扩增circRNA, 引物的方向应该是Divergent 方向。
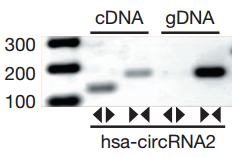
通过电泳的结果可以看到,对于cDNA , 使用Divergent 方向的引物可以扩增出circRNA 片段,而对于基因组DNA(gDNA), 则扩增不出对应的片段来。
二、find_cri使用流程
1) 基因组建索引:bowtie2-build genome.fa genome.fa(bowtie2v2.2.1)。
2) 安装python包:numpy (http://www.numpy.org/)和pysam (https://pypi.python.org/pypi/pysam)。
3) 建立一个文件夹genome,里面存放基因组中各个染色体的fasta文件,文件命名需同基因组fasta文件里的id相同。
4) 将处理后的读段比对到参考基因组上并将结果排序和转换成BAM格式:
bowtie2 –p 20 - -very-sensitive --score-min=C,-15,0 –q –x genome.fa -1 sample_1.fastq -2 sample_2.fastq 2>log/sam- ple.bowtie2.log| samtools view -hbuS - |samtools sort – sample
5) 取出未匹配上的读段:samtools view -hf 4 sample.bam|samtools view -Sb -> BAM/unmapped_sample. bam。
6) 将未匹配的读段两端各取20 bp作为锚点序列:python unmapped2anchors.py BAM/unmapped_ sample.bam|gzip>gzip/sample_anchors.qfa.gz。