服务热线02152235399
一、CIRI简介:
CIRI 根据circRNA 连接点处的reads来识别circRNA, 在连接点处的reads 其比对情况非常特殊;
CIRI 根据3种模型来识别circRNA, 连接点处的read 叫做junction read
A)
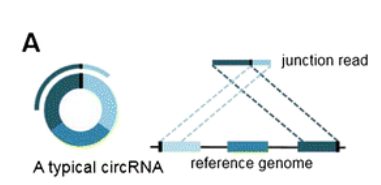
circRNA 由3个外显子环化形成, 由于测序读长的限制,junction read 只覆盖了起始外显子和终止外显子的部分序列,这两部分reads的比对位置在基因组上的位置是相反的
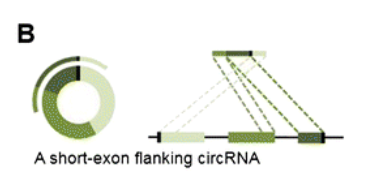
circRNA 由3个外显子环化形成, 由于连接点处的一个外显子其长度太短,junction read 除了覆盖了起始外显子和终止外显子的两部分序列外,还覆盖了中间的一个外显子的部分序列
C)
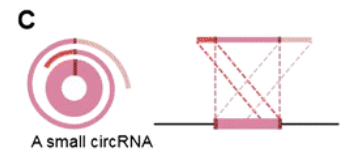
circRNA 由1个外显子环化形成, junction read 除了覆盖了整个外显子外,还重复又读了一部分序列
D)
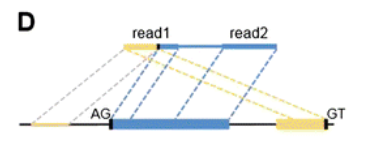
为了进一步降低假阳性率,CIRI 通过以下3条规则对结果进行过滤:
1)双端测序的两条reads 必须符合PEM 信号,以上面的示意图为例,进行说明read1 是一条junction read, 来源于两个外显子,根据read1 的比对情况,确定了circRNA 在基因组上的位置,此时,如果这个circRNA 识别准确,那么read2 就肯定落在对应的位置内;
根据两条reads的比对情况,进一步过滤结果;
2) 检测到的circRNA 的连接处符合AG-GT 剪切信号;
3)根据比对的质量和数量进行过滤,质量就是说mapping 的质量越高,识别的circRNA 越准确;数量就是说对于某个circRNA来说,检测到的juntion reads 越多,说明这个circRNA越可靠;
上面图中的几种模型只是帮助我们理解了exonic-circRNA的检测,其实对于non-exonic circRNA(包括intronic circRNA 和 intergenic circRNA)的检测,其原理是相似的,只是综合考虑了测序读长和连接点两段序列的长度,提出几种可能的比对模型,然后根据比对模型来检测对应的junction reads, 从而预测circRNA;
circRNA 结果的验证:
以一个预测得到的circRNA chr2: 58,311,224|58,316,858 为例,在基因组上的长度为 5634bp, 其连接点为VRK2基因的exon6和exon10
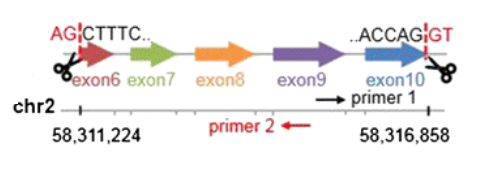
理论上产生的circRNA的序列为所有外显子组成的序列,splicing length为407bp
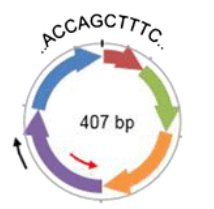
为了验证该circRNA , 根据连接点两端的序列设计引物,扩增出该circRNA 片段,跑电泳,确定产物长度

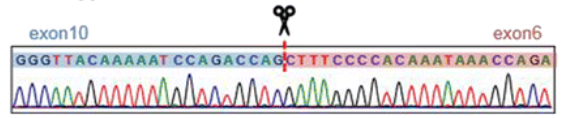
图中的黑色片段为扩增产物的条带,根据PAGE 电泳的结果,确定其长度;然后进行一代测序,确定具体序列
文献:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0571-3#Sec18
二、CIRI安装
2.1 下载地址:
https://sourceforge.net/projects/ciri/files/latest/download
2.2 安装方法:
解压即可。
三、CIRI使用方法
Usage: perl CIRI.pl -I in.sam -O output.ciri -F ref.fa (-R ref_dir/)
Arguments:
-I, --in
input SAM file name (required; generated by BWA-MEM)
-O, --out
output circRNA list name (required)
-F, --ref_file
FASTA file of all reference sequences. Please make sure this file is
the same one provided to BWA-MEM. Either this argument or
-R/--ref-dir is required.
-R, --ref_dir
directory of reference sequence(s). Please make sure fasta files in
this directory are from the FASTA file(s) provided to BWA-MEM. Either
this argument or -F/--ref-file is required.
-A, --anno
input GTF/GFF3 formatted annotation file name (optional)
-G, --log
output log file name (optional)
-H, --help
show this help information
-S, --max_span
max spanning distance of circRNAs (default: 200000)
-high, --high_strigency
use high strigency: only output circRNAs supported by more than 2
distinct PCC signals (default)
-low, --low_strigency
use low strigency: only output circRNAs supported by more than 2
junction reads
-0, --no_strigency
output all circRNAs regardless junction read or PCC signal counts
-U, --mapq_uni
set threshold for mappqing quality of each segment of junction reads
(default: 10; should be within [0,30])
-E, --rel_exp
set threshold for relative expression calculated based on counts of
junction reads and non-junction reads (optional: e.g. 0.1)
-M, --chrM
tell CIRI2 the ID of mitochondrion in reference file(s) (default:
chrM)
-T, --thread_num
set number of threads for parallel running (default: 1)
-Q, --quiet
keep quiet when running
-D, --output_all
keep the temporary files after running (more disk space would be
needed)
四、检测流程
1.使用BWA-MEM进行比对,
2.使用CIRI2进行检测,使用命令如:perl CIRI2.pl -I sample.sam -O test.ciri -F chr1.fa -D -Q -0 -S 200000 -A
CIRI 运行过程中所需要的内存资源比较多