服务热线02152235399
MutMap的分析方法如下图:
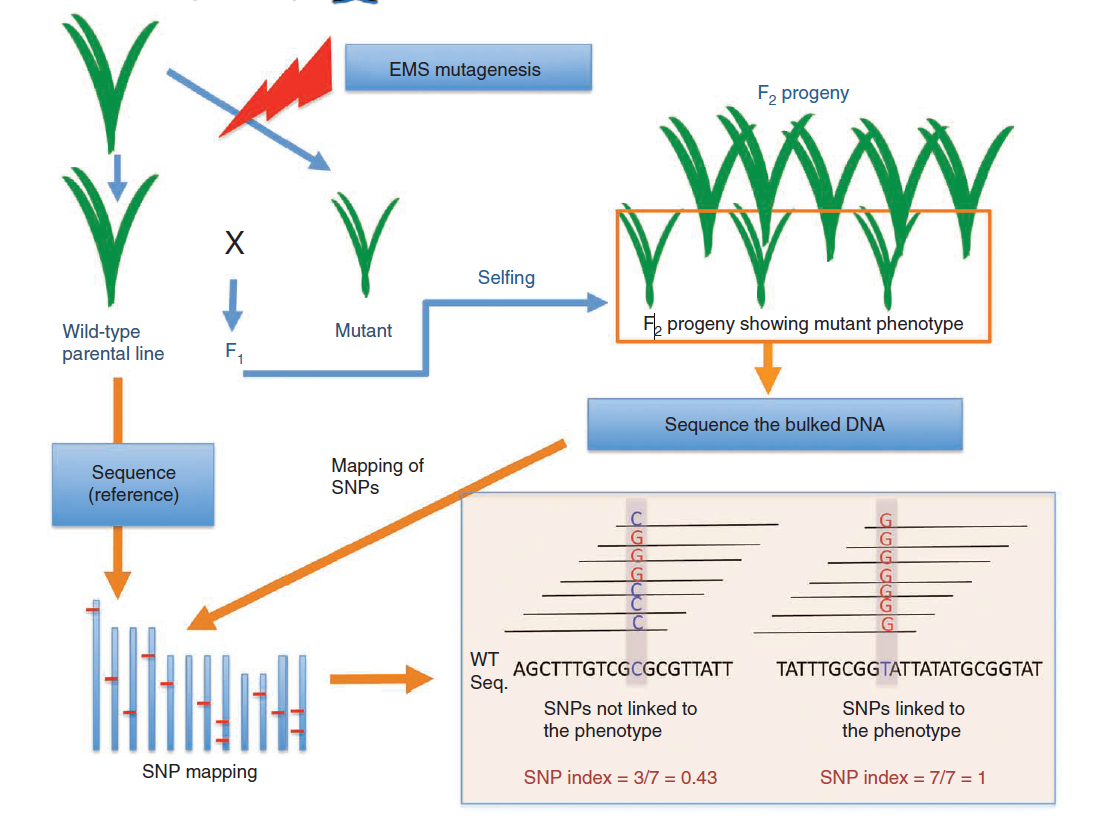
选择EMS处理的突变体进行回交获得F1子代,再自交获得形状分离的F2子代,选择F2中具有突变形状的植株进行混池测序,理论上与突变性状相关的突变率为1,而与突变性状不相关的突变率接近0.5,再绘制snp-index
与染色体位置的图,锁定峰值所在的区域进行验证,如下图:
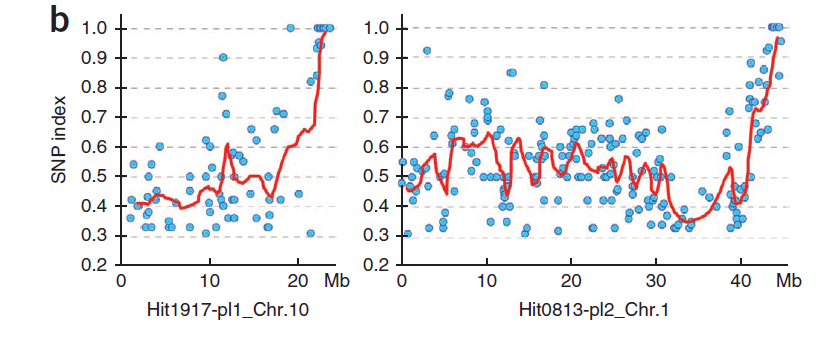
蓝色的点代表突变位点,红色的线是根据windows分析的结果绘制,每个window包含五个snp位点,纵坐标取突变率的平均值,横坐标取第一和第五的突变位置的中点
图中snp位点的筛选标准如下:
1.突变频率与覆盖度
纯合位点认为SNP-index大于等于0.9且覆盖度大于等于3
杂合位点认为SNP-index大于等于0.3且小于0.9,覆盖度大于4
2.去除不同样本间共有突变
去除掉至少有两个突变方向共有的SNP位点
3.根据EMS诱导突变原理筛选
由于EMS诱导的突变主要集中在G→A和C→T,所以图中只保留了这两种突变
在文章的附件中给出了一系列不同覆盖度与不同混样数量情况下,性状不相关突变位点在SNP-index上的频率分布图
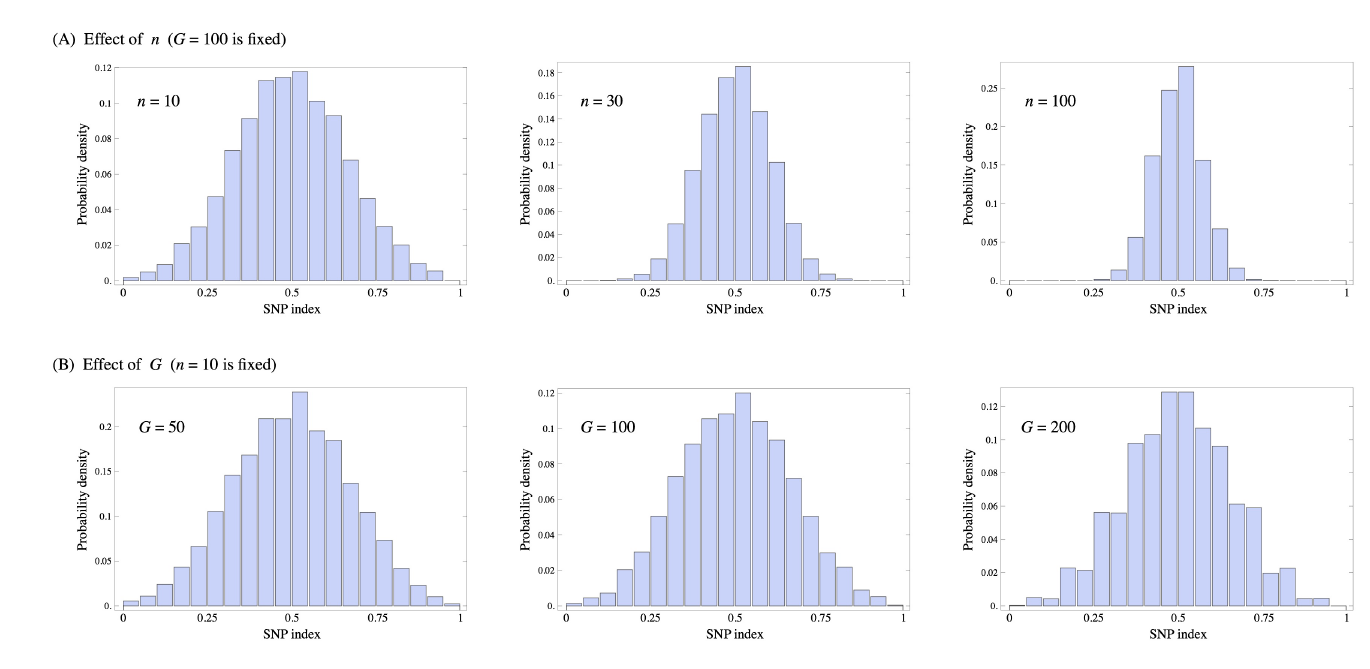
其中n代表混养池中样本的数量,G代表平均覆盖度,由图中来判断出纯合突变的SNP-index阈值
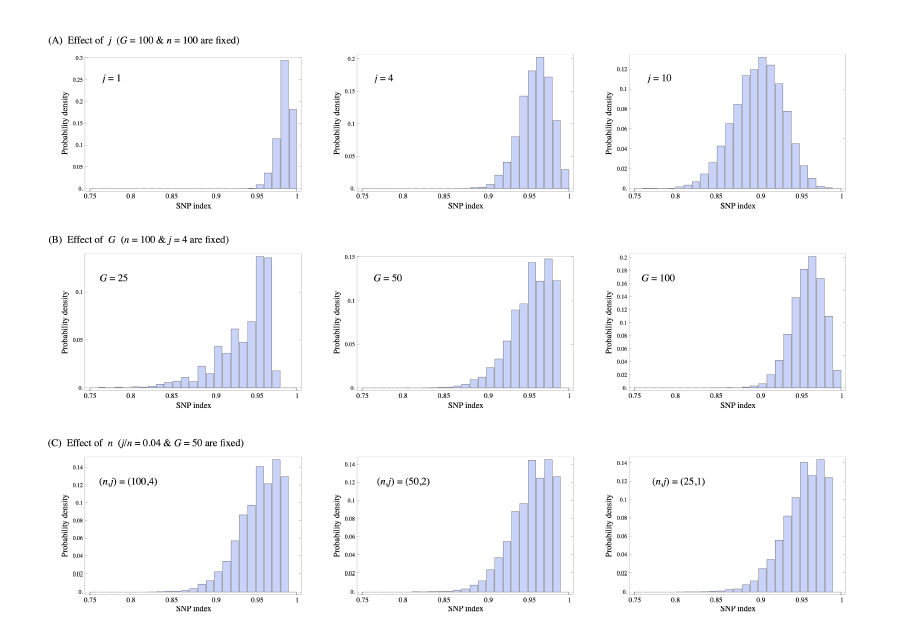
此外,mutmap方法还考虑了在突变株中存在极少量不突变的样本情况,也给出了在这种考虑下的与性状相关SNP在SNP-index上的频率分布图,如下:
其中j代表假设的未突变样本数,n代表混养池中样本的数量,G代表平均覆盖度
NovelBio实验室数据测试:
本次测试主要使用的测试样本为A,其中陈总给出的突变基因为XXX,所在染色体的位置为:XXXX
测试中使用Varscan算法对xxx与对照组9522样本进行callSNP,提取其中的somatic突变进行后续的分析,其中包含一个在目标基因上发生错义突变的位点,且突变率为1.
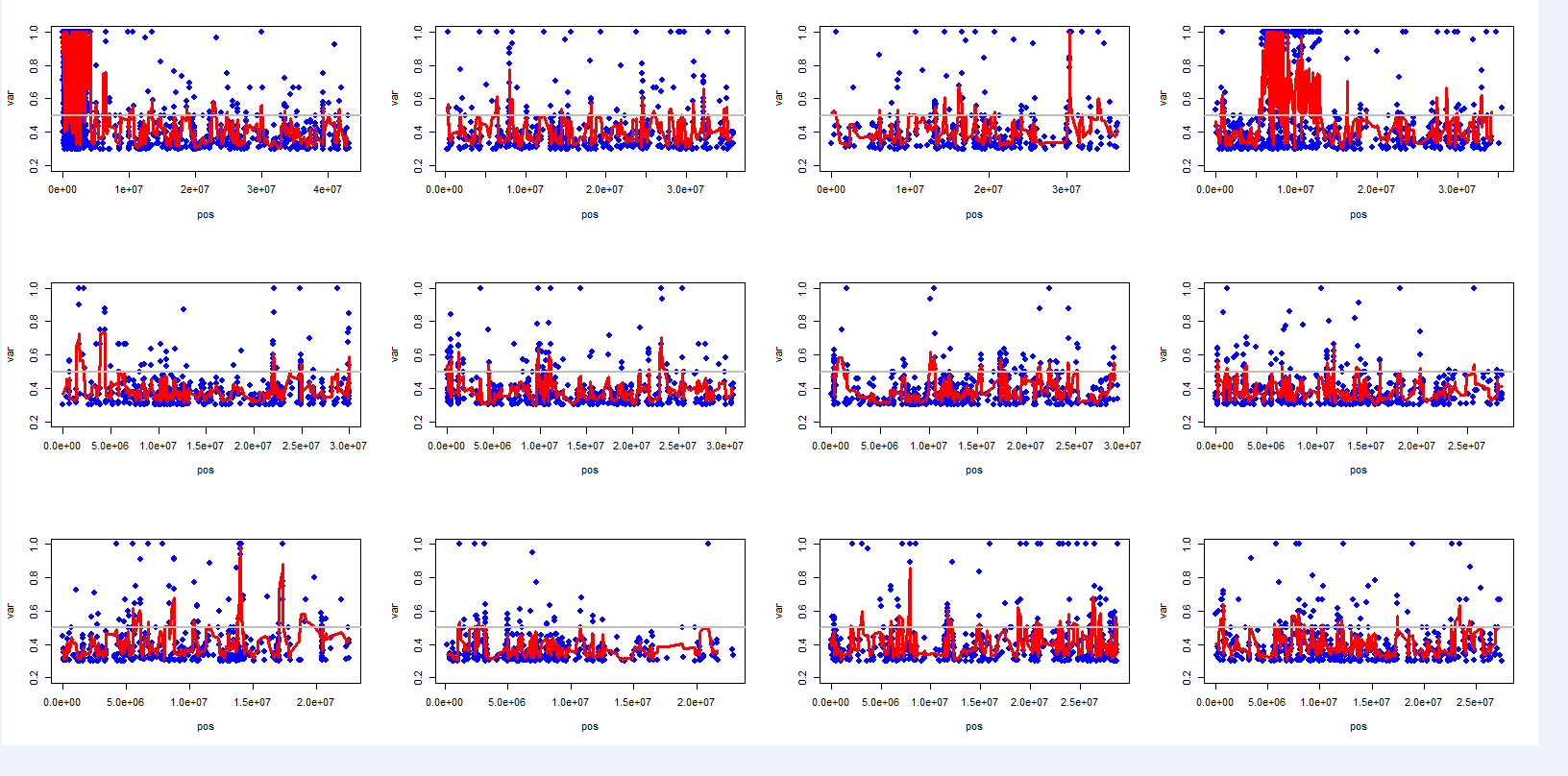
由图中可以看出具有高突变率的位点较多,在11号染色体关注基因区域没有明显的峰值,在第一,第四染色体有明显的突变富集,但是突变位点过于密集,经过IGV观察后看到如下情况:
1.目的基因处的突变位点为真,但是突变位点周围没有很多高频突变位点,导致图中没有明显的峰
2.在大量突变位点富集的区域,在对照组中也存在非常多的突变,导致结果不可信,而且在该位点存在过多的位点也使结果不是很可信
3.根据覆盖度的过滤会存在局限性,有部分位点可能同时存在多种突变类型,或者snp和indel共存的情况,这种位点也不是非常可信
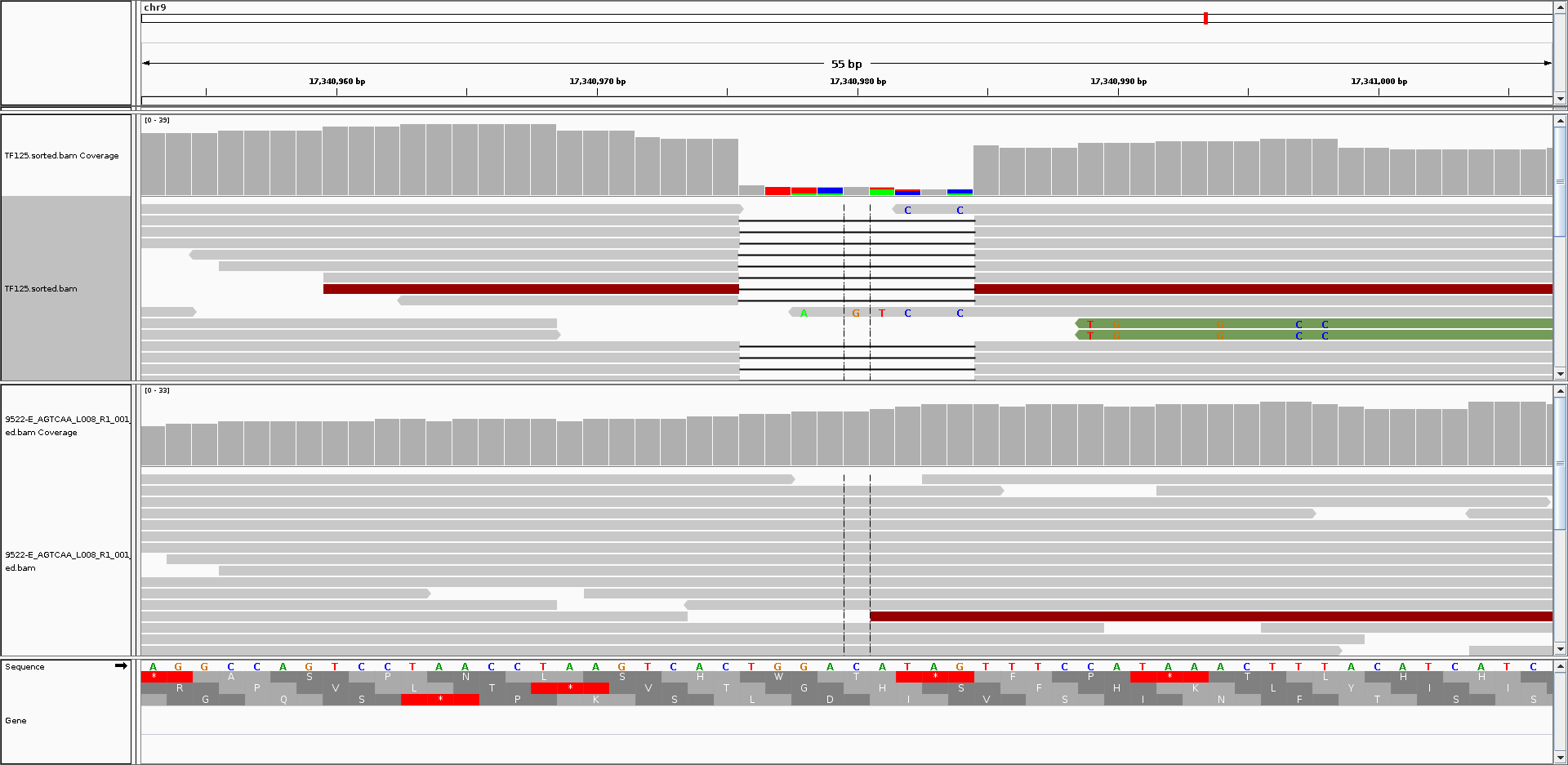
4.部分位点存在几个位置接近的纯合突变位点,图中存在不是非常明显峰的区域,但是不在基因的外显子区,未发生氨基酸的改变
初步结论,从图中来看,没有达到文章中出现明显峰的程度,考虑的原因是突变位点过多,产生的干扰比较严重,XXX这个样本的平均覆盖度为24,已经达到文章中提到的(>10×)的要求,但是画图使用的突变位点的数量相差一个数量级,对结果的影响很大。在Mutmap的文章中同时对一批F2子代的多种突变类型进行研究比如高矮,叶片颜色,并删除了很多共有突变(即可能不特异影响突变性状的位点),而且只考虑了G→A和C→T这两种突变类型,这就可以过滤掉非常多的位点。而在我们的测序数据中,只考虑了覆盖度的问题,在我对覆盖度梯度测试时,始终不能达到满意的结果,卡值过低,绘图所使用的突变位点会更多,干扰非常大,如果卡值过高又会丢掉非常多的信息。此外,由于陈总关注的目的基因所具有的突变类型为C→G,而且也不知道具体诱导突变的过程,没有对位点的突变种类进行筛选,这个也是导致突变位点很多的原因之一。
Q69样本补充分析,选择Q69的somatic突变位点进行过滤画图,过滤标准为tumor组覆盖度大于等于8,突变率大于30%,normal组突变率为0,只选择G→A和C→T这两种突变类型进行分析,
总共获得突变位点2173个,结果图片如下:
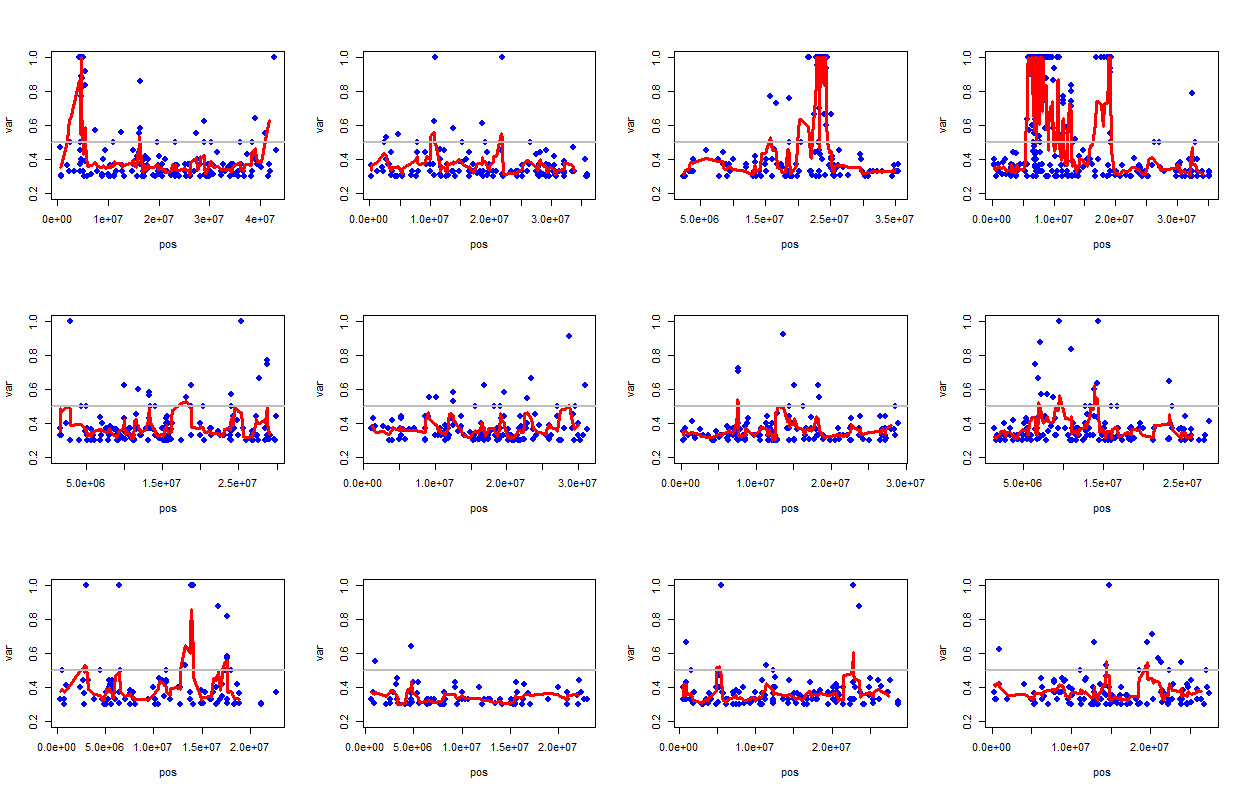
之后在对覆盖度的卡值进行梯度测试,由于在当时进行varscan分析时平台的tumor最低阈值为8,当卡值为8时获得2173个突变,当卡值为10时获得 当卡值为1085个,15时获得375个突变位点。