服务热线02152235399
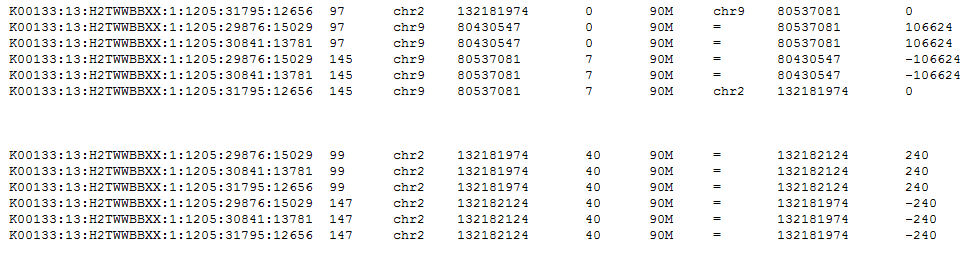
最近在分析一个重测序的SV过程中出现了一些问题,在使用lumpyExpression分析时,获得disordants Reads的时候出现异常,几乎获得的全部的mappingReads,原因探查过程如下:
提取disordants Reads的时候使用的方法是samtools,根据每一条reads的FLAG进行判断,具体代码如下:
samtools view -b -F 1294 sample.bam > sample.discordants.bam
-F代表过滤掉对应FLAG的reads,1294代表reads的FLAG情况,具体包含的reads如下,在这一步骤的核心是要过滤掉“read mapped in proper pair"的reads,即左右两端mapping在一致区域的reads

这个html为一个小程序,如上面的截图,输入Flag的编号可以得到对应哪些类型的Reads
在BWA mem的软件参数中包含一个 -P参数,具体介绍如下:
-P In the paired-end mode, perform SW to rescue missing hits only but do not try to find hits that fit a proper pair.
说明如果输入-P参数,在mapping的过程中或跳过fit a proper pair的步骤,也就意味着结果不会给出read mapped in proper pair这样的一个FLAG,而我们平台现在的BWA mem代码包含这样一个参数,如下:
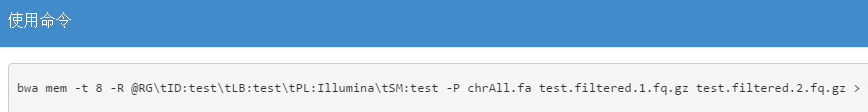
具体参数测试结果如下:
包含 -P 参数:
![]()
同样的的reads的FLAG变成了83和163,代表含义如下,包含read mapped in proper pair的注释
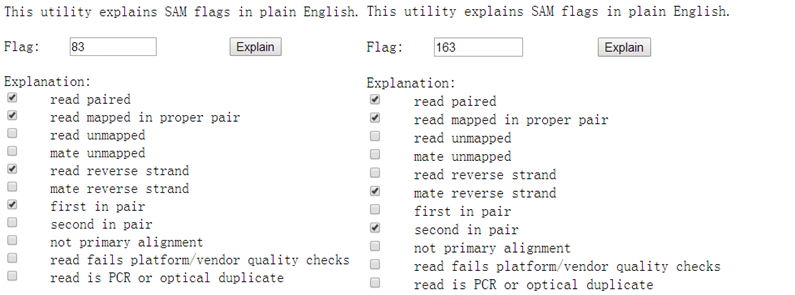
由此可见-P参数会影响每一条reads的FLAG,由于这种read mapped in proper pair在SV的分析过程中的判断是十分重要的,所以建议删除掉平台BWA mem模块的-P参数
经过测试,BWA -p参数除了影响FLAG外还会影响readsMapping的位置,在CallSNP过程中会造成很大的偏差,测试结果如下:
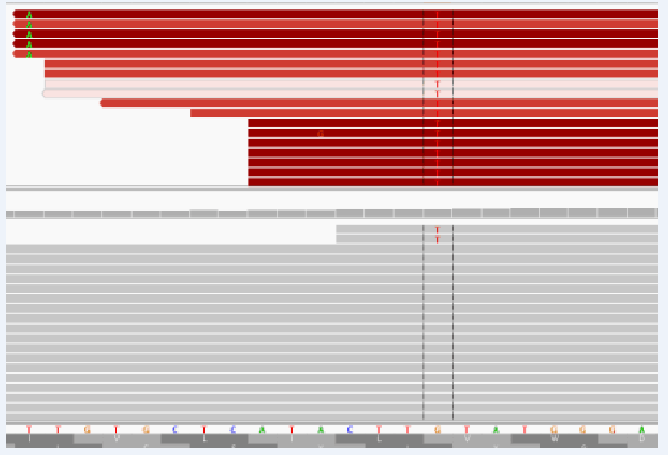
下图为同一条序列在不同mapping方法中的位置差异,上面为添加-p参数的结果,下面为不添加-p的结果,可以发现,不添加-p组中,每一条reads的左右两端均可以匹配在临近区段,且均在第二号染色体,而在添加-p组中,reads几乎全被mapping到9号染色体上,且左右两端reads的距离差距非常大,IGV截图(截图中为chr9对应区域,上面为添加-p参数,下面给为不添加-p参数)中也可以发现添加-p参数计算出的SNP位点存在明显的假阳性现象,再次证明-p参数需要被移除