服务热线02152235399
一 引言
1.1编写目的
进行该测试以及撰写此报告有以下几个目的
1.通过对测试结果的分析,得到对软件质量的评价;
2.分析在Illumina测序平台下,MapSplice能够获得最大junction数目以及mapping率的参数;
3.分析在ionproton测序平台下,MapSplice能够获得最大junction数目以及mapping率的参数;
4.尝试找到参数与测序长度的经验性关系。
1.2背景
MapSplice是一个RNA-seq数据分析工具,其核心程序是bowtie.可以快速的确认exon-exon剪切拼接。主要功能和Tophat差异不大。
与Tophat不同的是,MapSplice并没有针对某一种测序平台而开发,所以对于75bp以下的短序列以及75bp以上的长序列reads都可以使用。目前,全球最大的癌症研究项目TCGA(The Cancer Genome Atlas)正在主要推崇使用这个软件。
Ionproton属于二代测序中较新的平台,可以认为是二点五代测序平台,其测序长度平均在100个bp以上。目前我们公司使用的就是这个平台的进行二代测序分析。
鉴于之前使用Tophat进行参数优化以后发现结果并不是很理想,所以决定跟换软件进行测试,寻找更好的结果。因此,提出此次工作内容,探索更好的参数配置,提高mapping率以及junction数目。
1.3用户群
主要读者:公司研发部,公司管理人员。
其他读者:项目及销售相关人员。
1.4 数据对象:
|
Illumina数据 |
Ionproton数据 |
|
Illumina-low:liguanhu human |
Ionproton-low: congsongfeng human |
1.5 测试阶段
软件测试
1.6测试工具
Samtools version:0.1.18;
IGV version:2.3.18;
Awk;
1.7 参考资料
MapSplice userguide
Wang K, Singh D, Zeng Z, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery[J]. Nucleic acids research, 2010, 38(18): e178-e178.
Trapnell C, Pachter L, Salzberg S L. TopHat: discovering splice junctions with RNA-Seq[J]. Bioinformatics, 2009, 25(9): 1105-1111.
二 测试概要
关于MapSplice参数测试从2013年9月19日开始到2013年9月26日结束,共持续7天,一共25个测试用例。
主要测试内容如下:
1. 软件安装以及依赖性测试。
2. 文件分割以后查找junction数目以及不进行分割查找junction数目的差异大小,能否接受,为今后并行化文件回帖提供依据。
3. Segment参数进行优化工作。
4. 针对Illumina测序平台数据以及ionproton测序平台数据的mapping能力差异。
5. 简要测试MapSplice检测融合基因的能力
2.1工作计划进展
|
测试内容 |
计划开始时间 |
实际开始时间 |
计划完成时间 |
实际完成时间 |
工作完成情况 |
|
软件安装 |
2013年9月19日 |
2013年9月19日 |
2013年9月19日 |
2013年9月23日 |
本地安装受阻,服务器端安装正常。 |
|
软件依赖性查找 |
2013年9月24日 |
2013年9月24日 |
2013年9月24日 |
2013年9月24日 |
顺利 |
|
不同测序平台回帖能力 |
2013年9月24日 |
2013年9月24日 |
2013年9月24日 |
2013年9月24日 |
顺利 |
|
文件分割与否回帖差异 |
2013年9月25日 |
2013年9月25日 |
2013年9月25日 |
2013年9月25日 |
顺利 |
|
Segment参数优化 |
2013年9月26日 |
2013年9月26日 |
2013年9月26日 |
2013年9月26日 |
顺利 |
|
融合基因检测 |
2013年9月26日 |
2013年9月26日 |
2013年9月27日 |
2013年9月27日 |
顺利 |
2.2测试执行
此次测试严格按照项目计划和测试计划执行,按时完成了测试计划规定的测试对象的测试。针对测试计划制定规定的测试策略,依据测试计划和测试用例,将网络数据以及我们观测的关键参数进行了完整的测试。
2.3测试用例
2.3.1功能性
1.测试主要实现,包括较高的mapping率以及较多的junction数目。
2.测试junction数目与文件分割与否的相关性大小。
三 测试环境
3.1软硬件环境
|
硬件环境 |
服务器 |
|
硬件配置 |
CPU:Intel Xeon 2.66GHz *20 Memory:90GB HD:29TB |
|
软件配置 |
OS:Fedora release 14,Ubuntu 12.10 MapSplice 2.0.8 |
|
网络环境 |
100M LAN |
四 测试结果
4.1 软件安装
安装中,我们使用的软件版本是MapSplice 2.1.5。在本地进行测试的时候由于当时未知的软件依赖关系,并没有安装成功。软件提示报错为本地bowtie没有在系统中找到,于是在本地安装了与软件要求对应的bowtie 0.12.7 。本地可以使用bowtie,但是MapSplice仍然报这个错误,于是放弃在本地进行安装。在服务器的安装很顺利,很快就测试通过。
4.2文件分割mapping与未分割mapping
进行文件分割运行的最主要的考虑是为了尝试能否进行分布式的计算,所以我们在这一部分的工作中将文件分割成4份分开进行运算,然后将这4个文件运行出的junction数目相加比较与未分割情况下的junction数目差异。为了得到更加准确的效果,在本次测试中,我们使用了3个测序深度的ionproton测序平台得出的reads,分别是20万个reads,200万个reads以及整个文件(一共33926644个reads)进行分析。文件统一分割为4个文件。测试结果如下:
|
|
nonsplit |
split1 |
split2 |
split3 |
split4 |
差值 |
Ration(%) |
|
junction数 |
18601 |
4891 |
4356 |
4306 |
4029 |
1019 |
5.4782 |
表 20万个reads运行所得结果及差值
|
|
nonsplit |
split1 |
split2 |
split3 |
split4 |
差值 |
Ratio(%) |
|
junction数 |
241668 |
53000 |
62933 |
49905 |
60191 |
15639 |
6.47127 |
表 200万个reads运行所的结果及差值
|
|
nonsplit |
split1 |
split2 |
split3 |
split4 |
差值 |
Ratio(%) |
|
junction数 |
4965976 |
1093096 |
1040558 |
1288306 |
1192253 |
351763 |
7.083462 |
表 所有reads运行所有的结果及差值
通过上述结果可以知道分开以后与未分开时相差大约5%以上(占未分开的junction数目)所以可以认为并不是很适合将reads分开以后进行mampping。
4.3 segment参数优化
由于在Tophat参数探索的过程中,我们通过分析发现在所有参数中,segment_length是对junction影响最显著的参数,所以我们在对MapSplice进行分析时,主要也是分析这个参数。在测试过程中我们发现当这个参数大于30的时候就会报错,而软件参数中segment_length的下限值限定为18,软件说明中推荐对于50bp以上的reads文件,建议使用25这个长度,而根据文献中算法设计的思路可以知道,当这个数据越大的时候,整个junction的敏感度就会越低,而对应的程序运行时间就会越短,与之相反,当这个数据越小的时候,整个junction的敏感度就越高,而对应程序的运行时间就会越长。在本次测试中,我们对这个数值从18到28进行了抽样实验,实验结果如下:
|
segment_length参数测试 |
|
|
|
|
|
|
seg_length |
18 |
19 |
20 |
22 |
28 |
|
junction_numbers |
63291 |
63498 |
62610 |
60430 |
56040 |
|
threads |
13 |
13 |
20 |
13 |
13 |
|
time |
1:07:45 |
31:15 |
13:15 |
20:54 |
12:12 |
|
raito |
12.6582 |
12.6996 |
12.522 |
12.086 |
11.208 |
|
time_per_thread |
312.6923 |
144.7692308 |
39.75 |
96.46154 |
56.30769 |
|
|
|
|
|
|
|
|
按照>10KB都过滤掉 |
|
|
|
|
|
|
|
18 |
19 |
20 |
22 |
28 |
|
junctions_filted |
58266 |
58359 |
57624 |
55556 |
51448 |
|
junction_numbers |
63291 |
63498 |
62610 |
60430 |
56040 |
|
conseved_ratio |
92.06048 |
91.90683171 |
92.03642 |
91.93447 |
91.80585 |
|
ratio |
11.6532 |
11.6718 |
11.5248 |
11.1112 |
10.2896 |
|
|
|
|
|
|
|
|
按照<20bp的都过滤掉 |
|
|
|
|
|
|
|
18 |
19 |
20 |
22 |
28 |
|
junctions_filted |
63269 |
63482 |
62589 |
60412 |
56023 |
|
junction_numbers |
63291 |
63498 |
62610 |
60430 |
56040 |
|
conseved_ratio |
99.96524 |
99.97480236 |
99.96646 |
99.97021 |
99.96966 |
|
ratio |
12.6538 |
12.6964 |
12.5178 |
12.0824 |
11.2046 |
|
|
|
|
|
|
|
|
按照<20bp以及>10Kb的都过滤掉 |
|
|
|
|
|
|
|
18 |
19 |
20 |
22 |
28 |
|
junctions_filted |
58244 |
58343 |
57603 |
55538 |
51431 |
|
junction_numbers |
63291 |
63498 |
62610 |
60430 |
56040 |
|
conseved_ratio |
0.920257 |
0.918816341 |
0.920029 |
0.919047 |
0.917755 |
|
ratio |
11.6488 |
11.6686 |
11.5206 |
11.1076 |
10.2862 |
|
|
|
|
|
|
|
|
mapping_ratio |
80.75 |
79.9 |
78.08 |
73.94 |
75.52 |
|
|
|
|
|
|
|
|
real_junction_ratio |
14.42576 |
14.60400501 |
14.75487 |
15.02245 |
13.6205 |
表 segment_length数值与junction数目关系表(以上ratio省略%)
从上述表格中可以看出对于不同的segment_length而言,junction数目的百分比确实是有变化的,总体趋势是segment_length越长,junction的数目就越少,由于RNA-seq回帖率与测序深度正相关的关系,我们可以推测对于更多数目的数据而言,这个数值会有提高。在数据记录中,我们同时也记录了任务运行的总时间,与文献符合的是,segment_length长度越短,运行时间就会越低,而且我们发现时间增长的速度是很夸张的。当segment_length为28的时候,运行时间是12分钟12秒,而当segment_length为18的时候,运行时间是1小时7分45秒。可以看出这个时间差是很大的。综考虑我们认为如果有需要,取20到22都是不错的选择。
4.4融合基因检测参数测试
本实验中,我们主要检测了检测融合基因以及检测junction之间的关系。我们的检测了在寻找融合基因情况下,junction数目的变化,全部结果如下所示:
|
指定参数non_canonical_fusion |
500000 |
|
|
|
|
|
seg_length |
18 |
19 |
20 |
22 |
28 |
|
junction_numbers |
39786 |
40269 |
40203 |
39450 |
未测试 |
|
threads |
20 |
20 |
23 |
20 |
未测试 |
|
time(min) |
|
|
13:34 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
按照>10KB都过滤掉 |
|
|
|
|
|
|
|
18 |
19 |
20 |
22 |
28 |
|
junctions_filted |
36952 |
37399 |
37361 |
36621 |
未测试 |
|
junction_numbers |
39786 |
40269 |
40203 |
39450 |
未测试 |
|
conseved_ratio |
92.87689 |
92.87293 |
92.93088 |
92.8289 |
|
|
ratio |
7.3904 |
7.4798 |
7.4722 |
7.3242 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
按照<20bp的都过滤掉 |
|
|
|
|
|
|
|
18 |
19 |
20 |
22 |
28 |
|
junctions_filted |
39775 |
40257 |
40191 |
39440 |
未测试 |
|
junction_numbers |
39786 |
40269 |
40203 |
39450 |
未测试 |
|
conseved_ratio |
99.97235 |
99.9702 |
99.97015 |
99.97465 |
|
|
ratio |
7.955 |
8.0514 |
8.0382 |
7.888 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
按照<20bp以及>10Kb的都过滤掉 |
|
|
|
|
|
|
|
18 |
19 |
20 |
22 |
28 |
|
junctions_filted |
36941 |
37387 |
37349 |
36611 |
未测试 |
|
junction_numbers |
39786 |
40269 |
40203 |
39450 |
未测试 |
|
conseved_ratio |
92.84924 |
92.84313 |
92.90103 |
92.80355 |
|
|
ratio |
7.3882 |
7.4774 |
7.4698 |
7.3222 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
mapping_ratio |
41.7 |
41.8 |
41.54 |
40.92 |
未测试 |
|
|
|
|
|
|
|
|
real_junction_ratio |
17.71751 |
17.88852 |
17.98219 |
17.89394 |
未测试 |
由于之前的测试,我们考虑的参数中已经放弃了segment_length等于28这个参数,所以在这一步中,为了节约计算资源,我们并没有计算segment_length等于28情况下的测试数据。从上表中可以很明显的看出当检测融合基因时,整体数据的mapping率明显下降。因此导致的real_junction_ratio数目的提升并不能认为可能是真的提升。
五.测试结论与讨论
5.1平台差异
通过查阅已经有的资料,我们知道Illumina测序平台和ionproton平台最直观的差别在于后者的平均测序长度比前者长;在我们测试的例子中,Illumina的测序长度在50-97个bp之间,而ionproton的测序长度在50到235个bp之间。从此可以看出两者的最合适参数应该是有差别的。通过上一次tophat与这一次MapSplice的比较,我们发现,无论如何提高tophat的参数,我们都很难接近MapSplice使用默认参数下的junction数目,所以我们认为对于公司ionproton测序平台,我们使用MapSplice会更加适合。而在我们的测试结果中,对于Illumina测序平台测试时,进行单端实验的结果如下:
|
|
ionproton_low |
Illumina_low_single_end |
|
Junction ratio |
17.80055013 |
3.478367662 |
表 ionproton与Illumina单端结果计算junction百分数
5.2文件分割测试
通过这个测试的结果,我们可以看出分割前后运行得出的junction数目差距为5%(相比未分割的情况)以上,并且这个数目随着我们的测序深度的提高而提高。所以从这个结果而言,我们认为不适合将文件分割进行处理。
5.3segment参数测试结果
在测试实验中,我们发现segment_length参数从28向18变化的过程中,总体趋势是由少变多变少,整体趋势图如下:
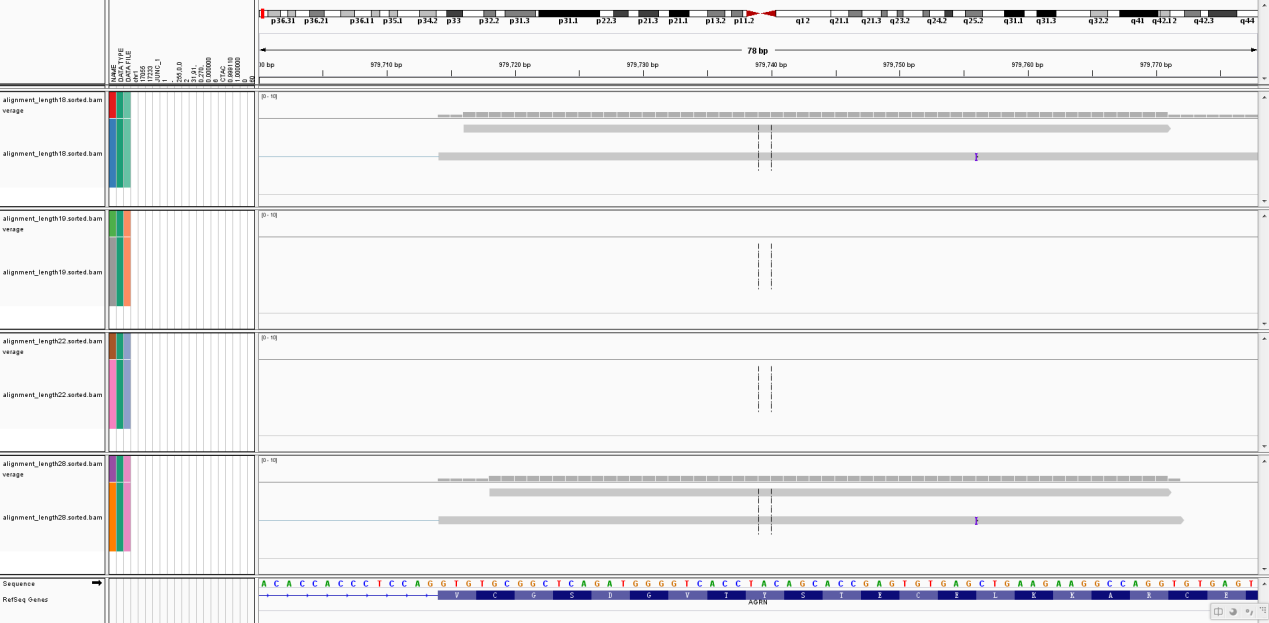
图 segment_length与junction_ratio关系图
从上图中可以看出大约在20到22的时候是最好的。在官方说明文档中,作者推荐当序列长度大于50的时候推荐使用参数25。下表是segment_length与测试时间之间的关系:
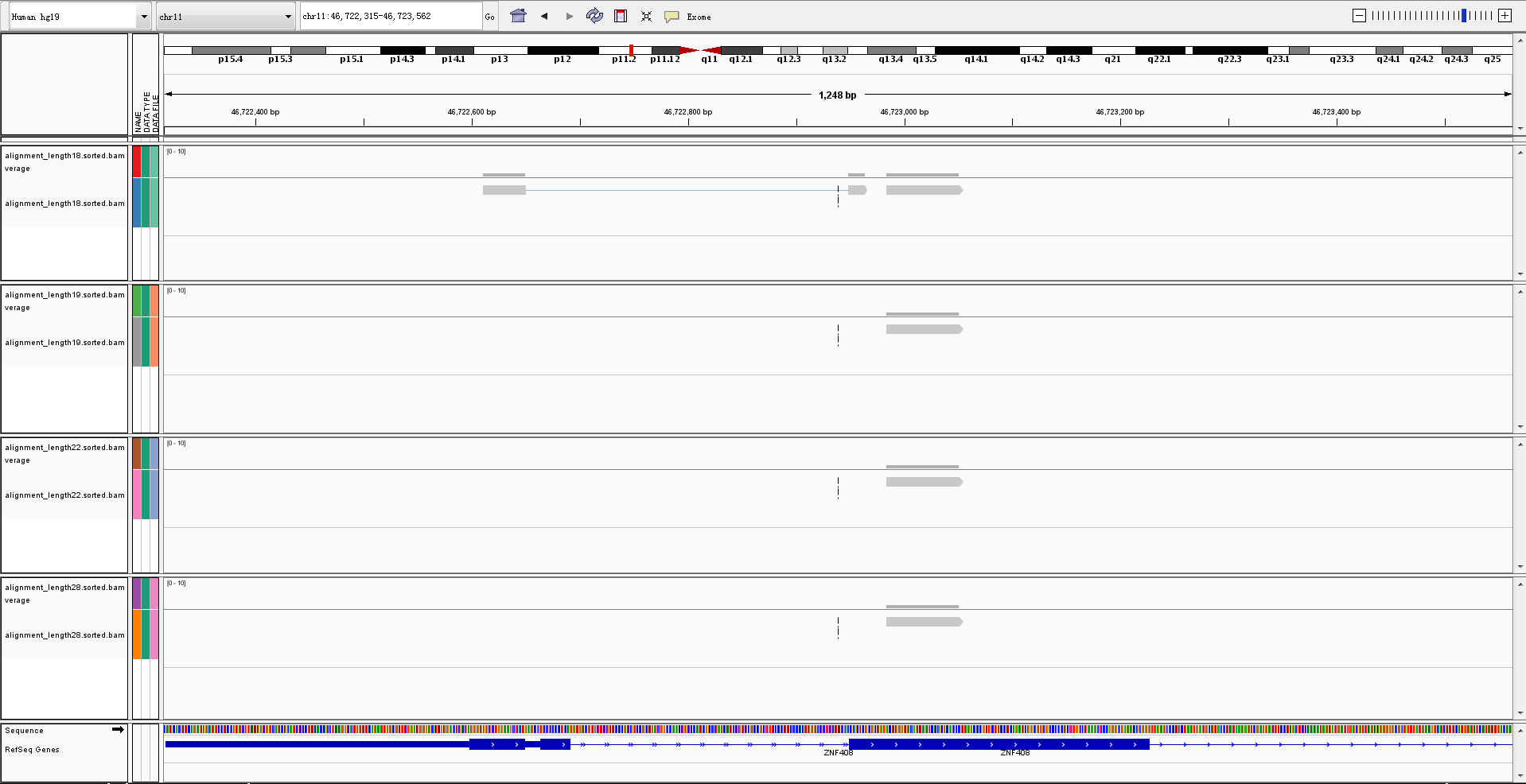
图 segment_length与running_time关系图
上图中,我们计算时间是使用实际计算总时间乘以运行的CPU数目。其中在22这个长度上时运行的CPU数目是20个,所以时间有所波动,总体而言来看,在长度为20到28之间时间变化还是可以接受的,然后当长度继续下降的时候,时间就开始指数级的上升的,这一点可以从图中看出。
所以,综合取舍junction率以及运行时间,我们认为使用默认参数是可以接受的,但是使用20至22也许会有更好的结果。
5.4融合基因检测参数设置
在我们的测试数据中,我们可以很明显的看出在各个segment_lengh情况下,mapping率都有下降,相比不做这一步检测,mapping率下降了至少30%,我们一开始认为是把部分junction的数据被认为是融合基因,当我们检测的时候才发现实际情况与我们的预测是不符合的。软件找到的融合基因数目十分少,并且基本都是跨染色体的。因此我们提出了新的想法,程序在同时进行查找junction以及融合基因的时候,为了确保计算时间不会超过单查找junction时的时间太多,并且由于查找融合基因是比较消耗计算资源的,所以程序在查找junction的时候并没有分配过多的资源,导致了更多的reads没有被程序mapping上去,因此我们在此认为实际应用中,应该将查找融合基因以及查找junction分成两步分开进行,如何能够使得两步的资源能更加节省,将是我们接下来的工作。
5.5 测试中的问题
在测试过程中我们发现了一个有趣的情况,如下图所示:
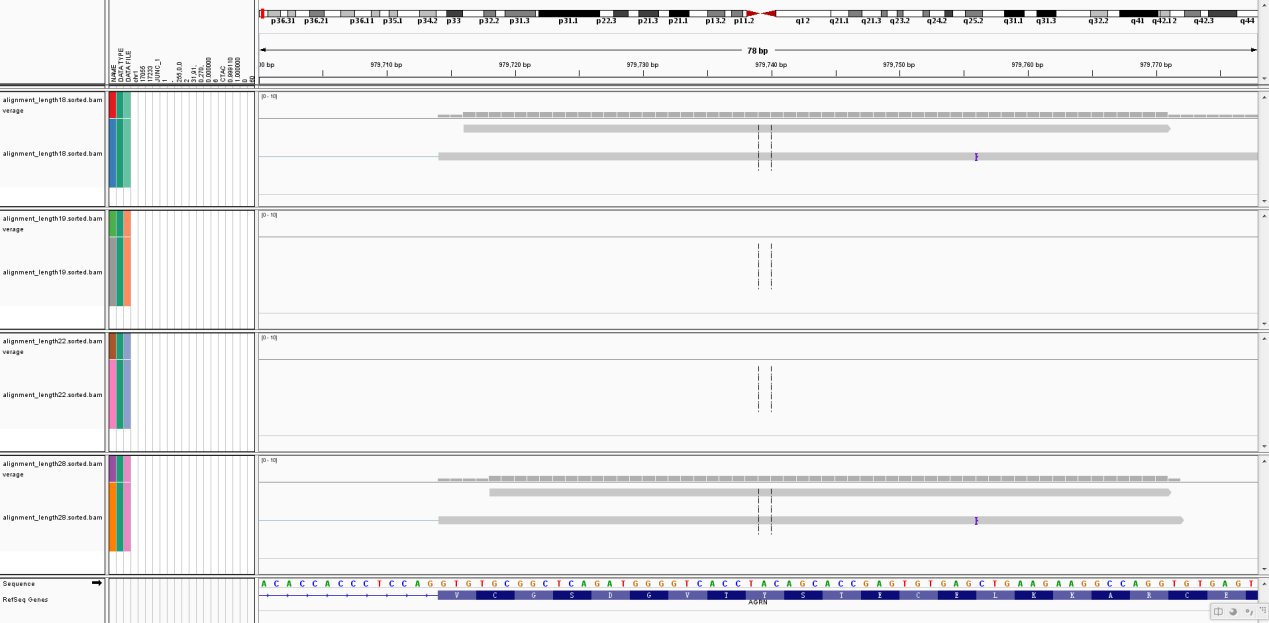
图 不同segment参数下查找junction数目的能力
上图中,首先可以看到在参考基因组中这个部分是有junction的,中间的四个条带从上到下依次是长度为18,19,22,28四个参数情况下的对应这个位置的回帖情况,可以很清楚的看到,在参数为18,28的时候是找到了这个区域的,但是在中间参数19,22的情况下,并没有回帖到这个位置,由于这个部分并不是很短,我们可以认为这个部分在染色体上是唯一的,所以排除了这两个参数情况下回帖到其他地方的可能性,确定这个部分对应的reads并没有回帖上去。因为并不是当segment_length变小或变大的情况下才逐渐出现的,所以可以认为是随机的结果,这暗示我们如果某一次的结果不是很理想的情况下可以通过重复或更改参数重复来提高junction数目。
另外在测试中,我们找到了支持segment_length越短,查找的敏感性就越明显的图像证据,如下图所示:
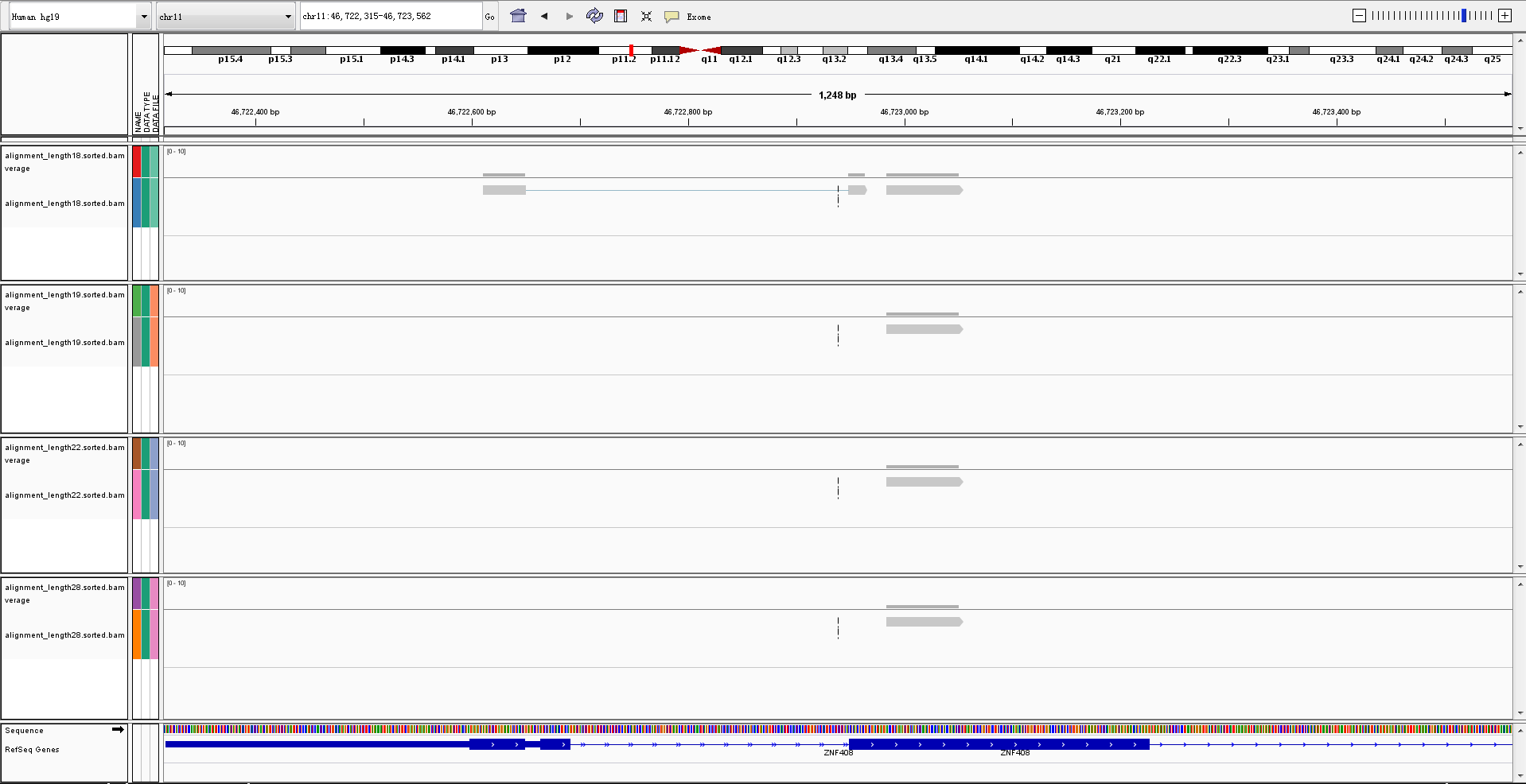
图 不同segment_length下查找junction的敏感性
上图中可以看出,在参考基因中,这个部分是有junction的,而在segment_length为19,22,28的时候,都没有找到回帖上,我们认为这个结果对于文献中提到的segment_length越短,敏感性越强这个说法。
六.测试总结
1.由于MapSplice在我们已经配置好的服务器上能够很流畅的直接使用,所以对于我们的hdfs而言,我们认为可以直接装配使用。对于本地的软件使用的可能需要复杂的软件支持,由于在这一步我们花费了部分的时间,所以在此并没有进行详细的寻找软件依赖关系。
2.综合考虑junction查找能力以及运行时间,我们认为在一般情况下,默认参数就是可以的了。当有特殊需求时,可以考虑使用参数在20到22内的任意值。
3.对于Illumina的单端数据而言,我们认为使用Tophat的效果比使用MapSplice的效果好,对于Illumina的双端数据而言, 对于ionproton的数据而言,我们认为使用MapSplice的效果远比使用Tophat好,不论是mapping率还是junction数都显示使用MapSplice更加合适。
4.我们认为查找junction以及查找融合基因这两个工作应该分开进行。由于时间关系,我们并没有查找弱化junction数目查找情况下,对融合基因查找的影响。
5.鉴于MapSplice查找junction时存在一定几率不能找全所有的junction,所以对于查找情况不好的数据,我们可以通过简单的重复运行或更改参数运行来尝试提高这个数据。
6.我们测试结果显示回帖操作并不能通过将源文件分割分别回帖来实现分布式运行。
七.测试中使用的命令,参数及说明
测试的结果在/media/hdfs/nbCloud/public/test/Illuminaandionproton0906/MapSplice-v2.1.5文件夹中下。
测试中统计junction数目的命令为
awk -F"\t" '{if((($3-$2)>20)&&(($3-$2)<10000)){total+=$5}}END{print total}' ./split4_test_segment_length20_non_canonical/junctions.txt
分割文件使用的perl文件见附件
使用MapSplice命令如下
Python MapSplice1.py -c /media/hdfs/nbCloud/public/nbcplatform/genome/human/hg19_GRCh37/ChromFa/sep \
-x /media/winE/genome/human/hg19_GRCh37/ChromFa/all/hg19_GRCh37_bowtie_index -o ./split4_test_segment_length25/ -1 ../split4.fq -p 10 -s 25
Mapsplice参数说明
其中重要的参数是粗体表现。
必须参数:
-c 序列文件的文件夹,注意:文件必须是fasta格式,后缀是.fa文件。
-x bowtie_index指定的路径及前缀。注意:只支持bowtie1的索引,并不支持bowtie2的索引。如果没有设定这个选项,或者指定的路径没有对应的索引,则会在结果输出路径下自动建立索引。
-1 FATSA格式或者是FASTQ格式。对于双端的回帖,这对应编号为1的文件。多个文件用逗号隔开,文件名之间不能有空格
-2 FATSA格式或者是FASTQ格式。对于双端的回帖,这对应编号为2的文件,并且两个文件顺序必须一致。多个文件用逗号隔开,文件名之间不能有空格
-p/--threads 线程数目,默认是1;
-o/--output 指定Mapsplice输出文件夹,默认是./mapsplice_out/这个文件夹 没有写清楚输出文件的具体样式,譬如输出文件前缀,文件名,输出文件类型
--qual-scale 输入文件的打分类型。默认是自动寻找,可以指定如下:phred33,phred64,solexa64
--bam 默认的输出文件时SAM格式的文件,通过这个选项可以指定输出BAM文件。
--keep-tmp 保存中间文件。
-s/--seglen 指定segment_length,通常默认是25,我们测试的结果暗示这个结果在20到22都是不错的。最小值是18,目前测试的结果暗示最大值不要超过30.
--min-map-len 软件只会记录完全匹配或者匹配数目不小于这个参数的序列。默认参数是50.
-k/--max-hits 每个read的最大匹配数,大于这个数的序列都丢弃掉。默认参数是4.
-i/--min-intron 最小intron长度,默认是50
-I/--max-intron 最大intron长度,默认是300000
--non-canonical 同样也搜索非经典的junction,我们测试的结果是这个参数能够提高junction数目,但是并不明显。
-m/--splice-mis 允许第一以及最后一个部分的最大不匹配数目。允许范围是0-2,默认参数是1。
--max-append-mis 允许匹配高出错率片段以及邻近的低出错片段不匹配的数目。默认参数是3
--ins 最大插入长度,默认是6,范围是0-10
--del 最大删除长度,默认是6,范围是0-10
--fusion| --fusion-non-canonical 查找融合(非经典)基因
--filtering junctions过滤级别,取值为1,2.默认是2;1代表更高的敏感度。2是标准过滤。