服务热线02152235399
清明假期马上就要到了,小伙伴们有没有做好出游计划呢~
我们都知道,清明节是中华民族数千年以来的重大春祭节日,扫墓祭祀,缅怀祖先。在这样一个寻根问祖的最好时节,作为生物医学领域的从业者,烈小冰也同大家一起在细胞层面追溯一下生物个体的起源,也算是应时应景。

今天烈小冰为大家介绍的文章由上海交通大学Bio-X实验室于2018年12月份以“Molecular characteristics of early-stage female germ cells revealed by RNA sequencing of low-input cells and analysis of genome-wide DNA methylation”为题发表于权威期刊DNA Research,文章引用了烈冰自主研发的可变剪接算法ASD(Alternative Splicing Detector,网址:http://www.novelbio.com/asd/ASD.html)烈冰技术团队也在其中提供了大量的数据分析服务和技术支持。


背景
在大多数多细胞生物中,包括哺乳动物,生殖细胞是一个新个体的起源,因此也承担着将遗传信息从上一代传递到下一代的职责。原始生殖细胞(Primordial germ cells,PGCs)是在发育初期阶段建立的第一个生殖细胞群[1]。PGCs在经历过迁移和性别分化之后,具有XX基因型的雌性胚胎PGC进入减数分裂的细胞前期I,然后在双线期阶段停滞,成为生发泡(germinal vesicle,GV)卵母细胞。在激素刺激下,GV卵母细胞完成第一次减数分裂,发育成为细胞中期II(MII)卵母细胞,又称次级卵母细胞。
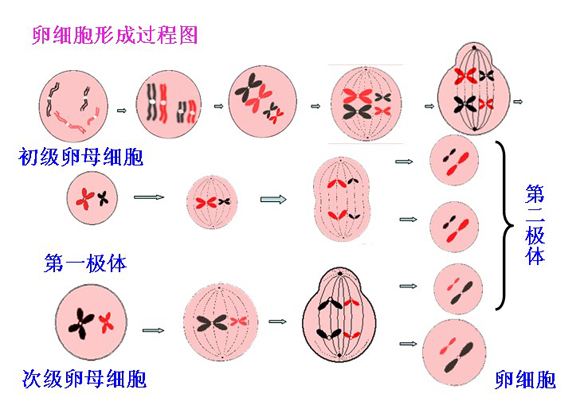
然而,最近的研究表明并非所有PGC都进入上述的分化途径[2-4]。研究报道,通过免疫磁性分选可以从5日龄和成年小鼠的卵巢中分离出雌性生殖系干细胞(female germline stem cell,FGSC)。在培养超过15个月后,FGSC仍然表现出增殖能力和正常核型,并且可以繁殖出可育后代[5]。另外,在产后的大鼠,猪和成年女性的卵巢中也证实了FGSCs的存在[6-8]。
该文章中,研究者使用RNA测序(RNA-seq)技术,对新鲜分离的FGSC的转录组和基因表达网络进行分析,并破解PGC,FGSC,GV和MII卵母细胞中基因表达的时空模式。这些研究对于更深入地了解雌性生殖细胞的发育过程和FGSC的分子特征至关重要。
文章思路:
1.分别对PGC,FGSC,GV和MII卵母细胞进行微量细胞的RNA-seq,获得表达谱数据;
2.通过对不同发育阶段的甲基化水平检测,对表达谱数据进行验证;
3.鉴定出与FGSC分化相关的关键pathway;
4.对不同发育阶段的基因表达进行加权基因共表达网络分析,鉴定出核心基因;
5.对不同阶段的lncRNA表达进行动态分析,鉴定出核心lncRNA;
6.对不同阶段的可变剪接模式进行分析,探索潜在分子机制。
结果展示:
1. 雌性生殖细胞在不同发育阶段的转录谱分析
研究者分别对PGC,FGSCs,GV和MII卵母细胞进行了5-8个细胞的RNA-seq,以检测不同阶段雌性生殖细胞中基因的表达情况,最终在37,980个已知小鼠基因中检测到平均17,805(47%)个基因的表达。特别是在FGSCs中,基因表达比例达到49%。为了探究这些基因表达谱是否与发育阶段相关,研究者使用非监督层次聚类算法来对RNA-seq数据进行分析。结果显示,这些雌性生殖细胞准确地符合发育顺序,从PGC到FGSC,并且如所预期的那样以GV和MII卵母细胞结束(图1A)。主成分分析(PCA)的结果揭示了发育阶段之间表达模式的差异(图1B))。所有阶段的基因表达的成对比较的结果也证明了阶段特异性的基因表达模式(图1C)。有趣的是,大多数差异表达基因(DEGs)可以聚集成反映阶段特异性表达模式的四个不同阶段,因此可能在雌性生殖系统发育过程中发挥关键作用(图1D)。
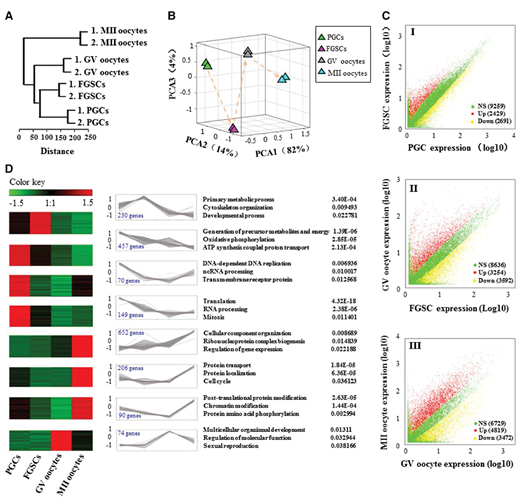
图1.不同阶段细胞的转录谱分析
2. 雌性生殖细胞在发育过程中的全基因组甲基化模式分析
为了进行雌性生殖细胞发育的全基因组DNA甲基化分析,研究者首先对新鲜分离的FGSC进行MeDIP-Seq,并分析了PGC, GV卵母细胞和MII卵母细胞的全基因组DNA甲基化的最新数据。结果表明,PGC具有相对较低的甲基化水平。性别分化后,FGSC也维持较低的甲基化,同时增加甲基化并分化成GV和MII卵母细胞(图2A),这与RNA-seq得到的基因表达数据形成呼应。为了明确新鲜分离的FGSCs与培养的FGSCs之间是否存在表观遗传模式改变,研究者对这两个群体进行了全基因组DNA甲基化分析,可以观察到二者之间显示出非常相似的DNA甲基化模式(图2B),并由染色体标度和Stra8基因座证实(图2C)。接下来,研究者分析了新鲜和培养的FGSC基因组中CpG岛(CGIs)的甲基化状态,发现培养的FGSCs中超过95%的甲基化CGIs可在新鲜的FGSC中被重新捕获(图2D))。对共享的甲基化CGI进行了功能注释,发现富集功能主要与体细胞发育有关(图2E)。总之,这些观察结果表明培养的FGSC的DNA甲基化模式几乎与新鲜FGSC的DNA甲基化模式相同。
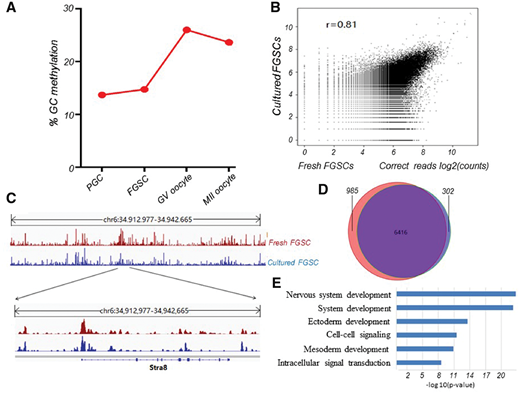
图2.不同发育阶段细胞的甲基化模式分析
3.雌性生殖细胞DEGs的pathway分析
为了探索雌性生殖细胞发育过程中涉及的生物过程相关pathway,研究者使用基于贝叶斯模型的聚类方法来分析从PGC到MII卵母细胞的DEGs,确定了13种重要的模型表达趋势(图3A),其中趋势1和10表现出从PGCs到MII卵母细胞的逐渐下降趋势,因此推断其与FGSCs的自我更新有关。相反,趋势8和24表现出从FGSC到GV卵母细胞的增加趋势,表明这两组与FGSC分化有关。有趣的是,趋势1/10共享一个途径PI3K-AKT(图3B I),而趋势8/24共享TGF-β途径(图3B II),提示这两种途径分别在维持自我更新和调节FGSCs分化中起着关键作用。 为了证实PI3K-AKT信号通路在FGSC自我更新和存活中的作用,研究者通过AKT抑制剂IV和染色实验证实AKT抑制剂IV处理的FGSC中凋亡细胞和死细胞的比例增加(图3C)。对于TGF-β途径,先前的研究将BMP4鉴定为PGC分化的关键调节剂[32],表明TGF-β超家族可能在小鼠卵子形成过程中调节FGSC分化,该发现与通路分析的结果相匹配,进一步验证了RNA-seq数据。
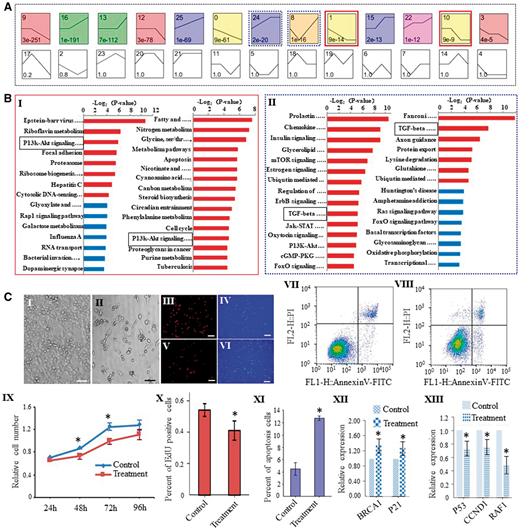
图3. 雌性生殖细胞DEGs的pathway分析
4. 雌性生殖系发育的加权基因共表达网络分析
为了研究雌性生殖细胞发育阶段与特定基因调控模块之间的共表达关系,研究者进行了加权基因共表达网络分析(WGCNA),发现雌性生殖系发育涉及25个共表达模块(图4A))。在类似功能模块合并后,获得了四个复合模块,这些模块显示了阶段特异性的表达模式。GO富集显示这些模块涉及一些阶段特异性生物过程,如RNA加工,细胞分裂和细胞周期(PGCs),有丝分裂细胞周期和核分裂(FGSCs),配子生成和有性生殖(GV卵母细胞),减数分裂染色体分离和程序性细胞死亡(MII卵母细胞)(图4B)。然后研究者检测了发育期间模块表达的分布,发现PGC模块在发育过程中逐渐降解,而从FGSC到GV卵母细胞或从GV到MII卵母细胞的转变中的模块显示出急剧退化和持续激活(图4C)。
研究者分析了模块和发育阶段之间的相关性(图4D),证明小鼠雌性生殖系发育也涉及阶段特异性共表达模块。接下来,为了鉴定核心阶段特异性调控基因,研究者构建了核心基因网络,并筛选出一组核心基因:Ncapd2,INCENP,Rab5b,PSAP,GTPBP3和EIF4H(图4E)。通过这些分析,研究者推测FGSCs可能与这些核心基因有关,并通过控制其有丝分裂进程进一步指导干细胞自我更新。
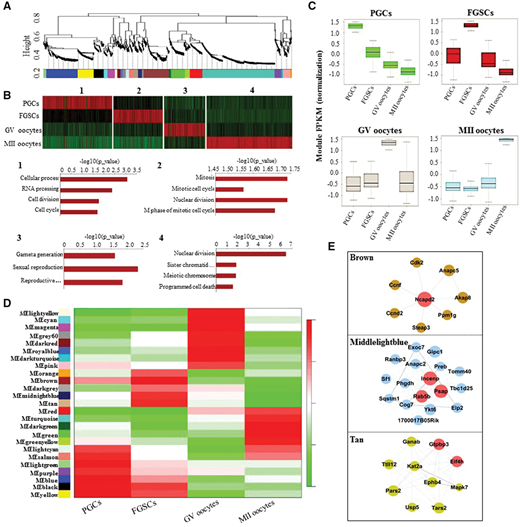
图4. 雌性生殖系发育的加权基因共表达网络分析
5.lncRNA的动态表达分析
为了追踪雌性生殖系发育中长非编码RNA(lncRNA)的表达,研究者鉴定出632个显著差异表达的lncRNA。经过层次聚类分析,研究者发现,与蛋白质编码基因一样,这些lncRNAs显示出非常独特的阶段特异性表达模式,表明它们在雌性种系发育过程中具有调节作用(图5A)。
miRNA是哺乳动物发育和体内平衡的重要调节剂,lncRNA通过与miRNA的结合调节其相应的mRNA。本研究中,研究者使用了三个核心基因(Ncapd2,CDKN1A和Rab5b),两个已知的生殖系标记基因(MVH和C-KIT)和一个干细胞相关基因(LHX1)构建交互网络,获得了功能特征性的lncRNA,包括XIST和MALAT1(图5B,C)。使用单细胞RT-PCR,研究者在FGSC阶段鉴定到XIST的表达(图6D),这与RNA-Seq数据一致。值得注意的是,研究者发现ZFP783可通过miR-6963-3p与FGSCs中的MVH和CDKN1A相互作用。由于CDKN1A已被证实对维持FGSCs很重要(图3C XII),因此揭示了ZFP783可通过miRNA海绵效应来对FGSC分化和有丝分裂细胞周期发挥调节作用。
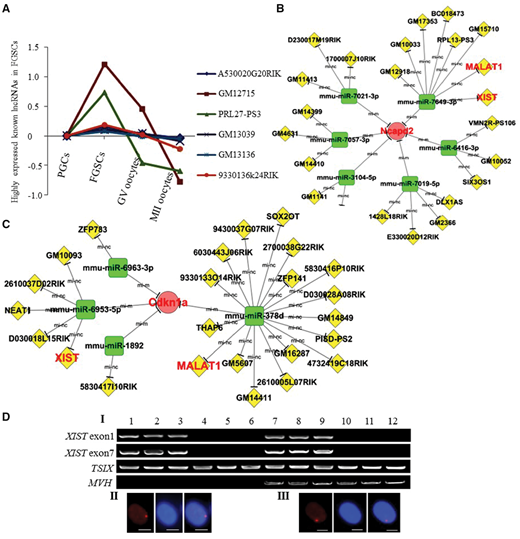
图5. lncRNA的动态表达分析
6.可变剪接的动态模式分析
在雌性生殖系发育过程中,研究者发现FGSCs的可变剪接(AS)发生频率最高(图6A),可能有助于FGSCs的自我更新和分化。在FGSC阶段具有两个或更多个转录物的已知基因的比例也显示在图6B中。
为了进一步研究FGSC特异性AS模式,研究者获得PGC与FGSC和FGSC与GV卵母细胞之间的AS的交叉元件用于后续分析。通过调整P值<0.05,研究者在FGSC阶段鉴定了698个AS事件(图6C),其中内含子保留和盒式外显子都是关键的AS模式。有趣的是,G蛋白偶联受体途径也参与到有丝分裂过程(图6D)。根据以往的研究,研究者绘制出几个核心基因(DTYMK,FHL1,CDK2,SYCP3和TBP)的AS类型(图6E)。综合这些结果,研究者推测动态AS模式可以影响有丝分裂相关的基因表达,继而促进FGSC的特性形成。
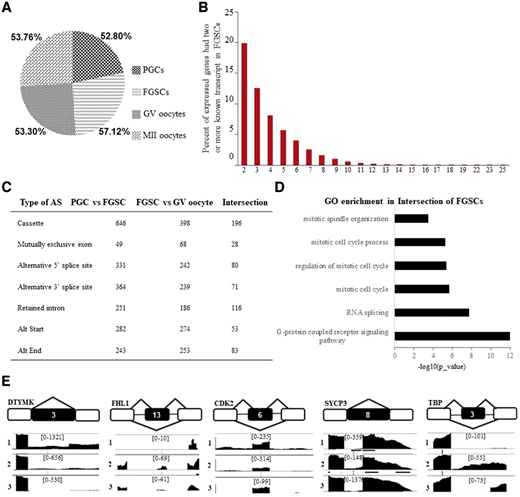
图6.可变剪接的动态模式分析
综上,本研究系统地分析了PGCs,FGSCs,GV和MII卵母细胞的形态和分子特征。通过RNA-Seq对已知的RefSeq基因,已知的lncRNA,新的lncRNA和AS的动态模式进行研究,描绘出雌性生殖系发育和FGSC来源的卵母细胞形成背后的发育阶段特异性和调节机制。这些发现对于研究卵细胞发育过程中的分子pathway和机制非常宝贵,并将在其他类型干细胞研究中得到更广泛应用。
烈冰会继续在自己擅长的生物信息分析领域深耕细作,推出更多更高质的产品和生信分析工具,凭借实力助力广大科研工作者的科学研究。想了解更多信息,请联系您的区域业务经理进行咨询~
参考文献:
1. Saitou, M. and Yamaji, M. 2012, Primordial germ cells in mice, Cold Spring Harb, Perspect. Biol., 4, 59–66.
2. Bukovsky, A., Gupta, S.K., Virant-Klun, I., et al. 2008, Study origin of germ cells and formation of new primary follicles in adult human and rat ovaries, Germline Stem Cells. Humana Press, 450, 233–65.
3. De Felici, M. and Barrios, F. 2013, Seeking the origin of female germline stem cells in the mammalian ovary, Reproduction, 146, R125–30.
4. White, Y.A.R., Woods, D.C., Takai, Y., et al. 2012, Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women, Nat. Med. , 18, 413–21.
5. Zou, K., Yuan, Z., Yang, Z., et al. 2009, Production of offspring from a germline stem cell line derived from neonatal ovaries, Nat. Cell Biol., 11, 631–6.
6. Zhou, L., Wang, L., Kang, J.X., et al. 2014, Production of fat-1 transgenic rats using a post-natal female germline stem cell line, Mol. Hum. Reprod., 20, 271–81.
7. Ding, X., Liu, G., Xu, B., et al. 2016, Human GV oocytes generated by mitotically active germ cells obtained from follicular aspirates, Sci. Rep.,6, 28218.
8. White, Y.A.R., Woods, D.C., Takai, Y., et al. 2012, Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women, Nat. Med. , 18, 413–21.